
- Messages
- 83
Been on MK7 for over a year and haven't noticed any difference.

What dosage you take?Been on MK7 for over a year and haven't noticed any difference.
What dosage you take?
I did not notice any difference either when I was taking 100mcg K2-MK4 + 500iu D3 in the same drop (Thorne).100mcg. I have taken it for my bones I never knew it may have a benefit to CFS.
Generally I get plaque accumulation between my lower teeth very quickly after visiting the dentist for dental cleanse, no matter how often I brush and floss. Today an entire piece of plaque just fell off. I am attributing it to eating 2 hard-boiled eggs (with runny yolks) daily, instead of just one as I used to do before... Or it is my current supplementation to lower oxalate burden:Effect on teeth yes.
They get very "clean", no more plaque. This is how I notice that I take enough MK-4
@Gondwanaland, is this a possibility with you?What happens with calcific tendinitis is that while the calcium deposits are building up in the tendon, it twinges a bit now and then. In the acute form, when the calcium is released, it goes nuclear, and even doctors call it "excruciating".
 . Thanks to everyone who's posted about their experiences. I'll sure post mine when I start trying K2.
. Thanks to everyone who's posted about their experiences. I'll sure post mine when I start trying K2.I am more inclined to think that K2 chelates the oxalates from the calcium in my bonesis this a possibility with you?
That is Ray Peat styleHas anyone tried putting the K2 drops on their skin?
He does recommend it transdermally From this page on the Weston A Price Foundation site....researchers from Tufts University published a paper in the Journal of Nutrition ... showing that vitamin A protects against vitamin D toxicity in part by helping to properly regulate the production of vitamin K-dependent proteins.
Apparently the liver increases fibrinogen production when vit K metabolism is disrupted by intake of blood thinners. My dr. asked for fibrinogen blood test back in June, but when my results came back above the range, she didn't know what to make out of themhttp://www.ncbi.nlm.nih.gov/pubmed/1489894
Blood Coagul Fibrinolysis. 1992 Dec;3(6):727-30.
Non-vitamin K-dependent clotting factors during oral anticoagulant treatment.
van Wersch JW1.
Author information
Abstract
The non-vitamin K-dependent coagulation factors fibrinogen, factor V, von Willebrand factor and factor XIII were measured in plasma of patients receiving oral anticoagulant treatment. Levels of fibrinogen, von Willebrand factor and factor XIII were above the upper normal limit in 60.0%, 58.6% and 53.4% of patients respectively. Factor V levels were below the lower normal limit in 56.3% of patients. Compared with a control group we found significant differences in the anticoagulated group for fibrinogen (P < 0.0001), factor V (P < 0.0001), von Willebrand factor (P < 0.0001) and factor XIII (P < 0.0001). It appears that the down-regulation of factors II, VII, IX and X during oral anticoagulation is accompanied by a subsequent up-regulation of fibrinogen, von Willebrand factor and factor XIII. This up-regulation or its absence could explain the thrombotic and bleeding complications seen in some patients on anticoagulant therapy and might also reduce the beneficial effect of oral anticoagulation in cardiovascular disease.
It took me some months of research, but I finally found out that Serrapeptase can help to reduce fibrinogen. I have started taking it this week and already noticed a decrease in inflammation I noticed I tolerate it better if I eat a bunch of high vit K foods (fresh herbs, leafy greens, pumpkin/sunflower seeds, tahini etc ). THEN I found the study above So have you taken anti-coagulants? Or is there some other reason your vit. K metabolism was disrupted?Apparently the liver increases fibrinogen production when vit K metabolism is disrupted by intake of blood thinners.
I took coumadin in 2011-2012, which worsened my general inflammation and LECTIN (gluten) intolerance horribly. I have found extensivwely documented connection between fibrinogen and lectins!So have you taken anti-coagulants?
Anyway, since I don't eat fish very frequently, I have started Beta-Alanine, which I think will be beneficial to fibrinogen-lectin-oxalate issues That's great! I stopped sweating for about 10 years after EBV. I think it came back about the time I started T3. Recently I have gone from sweating lightly to a bit more.I like how Serrapeptase raises my BP and makes me sweat! (I never sweat)
Well I do, and I'm feeling better without the krill oil I was taking, which I thought was weird; I started taking it after reading Russell Blaylock's book on brain health.Ray Peat is against Omega 3 ("turns your brain into mush"). Well, Inuits go thru winter thanks to Omega 3, but I don't live in a place with harsh winters
Hmmm, that's interesting, because my RBC count is slightly over the top of the range (5.17 X 10-6/uL), but it was at that level two years ago, before I was taking lots of Bs or Vit. K.I think that heavy supplementation with B vits + vit K can thicken the blood dangerously (> RBC count
That might explain it, since I have been taking B5 for years, but wasn't taking B6 at the time.Esp. B5 that shifts the balance into calcium retention and must be well balanced with B6 which holds magnesium.
my RBC count is slightly over the top of the range (5.17 X 10-6/uL),
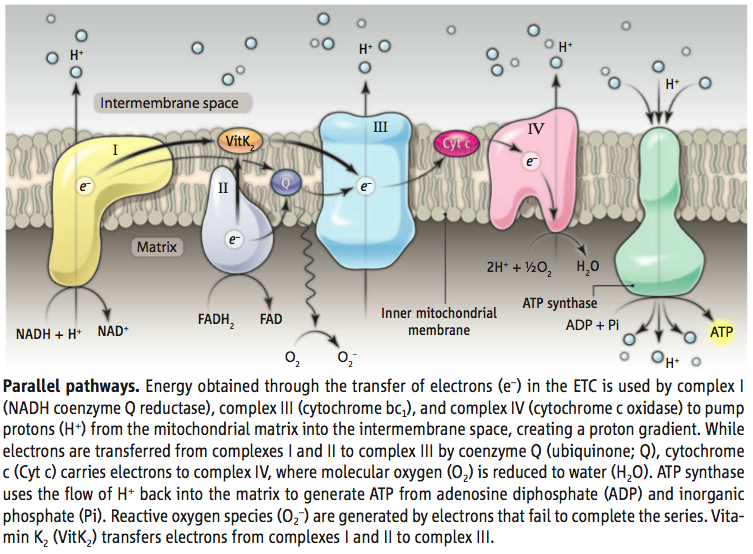
Mitochondria are dynamic organelles that play central roles in eukaryotic cellular energy metabolism. They harbor an electron transport chain (ETC) that couples electron transfer to the movement of protons across the mitochondrial inner membrane, forming an electrochemical gradient that captures chemical energy in the form of adenosine triphosphate (ATP). The biochemical and biophysical properties of the ETC have been studied in detail (1). On page 1306 of this issue, Vos et al. (2) report a new constituent of this chain. The authors show that vitamin K2 is an electron carrier, suggesting this small organic molecule as a possible treatment for mitochondrial pathologies such as Parkinson’s disease and amyotrophic lateral sclerosis.
In the ETC, a series of electron donors pass electrons to more electronegative acceptors until electrons are passed to oxygen, forming water (see the figure). A number of vitamins are involved in this relay. For example, nicotinamide (vitamin B3) is the precursor of NADH (the reduced form of nicotinamide adenine dinucleotide) and delivers electrons to the first protein complex in the ETC, complex I; riboflavin (vitamin B2) is the precursor of FADH2 (the reduced form of flavin adenine dinucleotide) and is a cofactor for electron transport.
Mitochondrial dysfunction has been linked to neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis (1, 3). In many of these diseases, some electrons react with oxygen prematurely, generating reactive oxygen species that damage cellular components. Causal links between mitochondrial dysfunction and neurodegeneration have been established, but the molecular mechanisms by which perturbing mitochondrial activity leads to synapse loss and neuronal degeneration are unknown.
Although our understanding of the etiology of Parkinson’s disease is incomplete, genes associated with rare, familial forms of the disease have been identified. Mouse and fruitfly models of the disease suggest that two genes, pink1 and parkin, function in mitochondrial quality control (4–8). Pink1 is a protein kinase that detects decreases in mitochondrial membrane potential and recruits cytoplasmic Parkin to damaged mitochondria (9). Parkin is an E3 ubiquitin ligase that directs the ubiquitin-proteasome system to degrade specific target proteins (10). Together, Pink1 and Parkin sequester damaged mitochondria prior to their clearance through a process called mitophagy (11, 12). However, these proteins likely have other functions that are as yet unknown.
Vos et al. screened a collection of mutant fruit flies with abnormal synaptic function for loci that enhanced or suppressed the flight defects seen in pink1 mutants. The screen identified heixuedian (heix), a gene that dominantly influences flight behavior, ATP levels, and mitochondrial activity in pink1 mutants. heix encodes an evolutionarily conserved enzyme involved in the synthesis of the quinone vitamin K2. Vitamin K2 has a well-established role in post-translational modification of proteins involved in blood coagulation (13); more intriguingly, it functions as an electron-carrier in prokaryotes(14).Vosetal. show that vitamin K2 functions as an electron carrier required for ATP production via the ETC in eukaryotic cells. Furthermore, supplementing the diet of pink1 mutant flies with vitamin K2 increased ETC efficiency and partially alleviated defects in mitochondrial membrane potential and ATP production.
A striking feature of this study is the uncoupling of defects in mitochondrial morphology from functional def icits. That is, although functional defects in pink1 and parkin mutant flies were alleviated by boosting ETC activity via rescue of heix function or by providing exogenous vitamin K2, these manipulations had a less dramatic effect on the defects in mitochondrial morphology observed in these animals. Also, although rescuing heix function or uptake of vitamin K2 in pink1 or parkin mutant flies provided functional rescue, whether Pink1 directly regulates Heix enzymatic activity is unknown. Def ining these interactions will provide insights into both mitochondrial biology and the function of Pink1.
It is tempting to speculate that evolution might have favored the emergence of parallel pathways for electron transport. Although these could simply provide “fail-safe” mechanisms for energy generation, the presence of parallel pathways could also hint at tissue-specific or cell type–specific specializations in the ETC. For neurodegenerative disorders in which there is mitochondrial dysfunction, each disease manifests in characteristic subpopulations of neurons. For example, in Parkinson’s disease there is relatively selective loss of dopaminergic neurons in the substantia nigra, whereas degeneration of motor neurons in the ventral horn of the spinal cord is prominent in amyotrophic lateral sclerosis (3). If ETC function is generic, why do these unique patterns of loss arise? The conventional explanation is that the specific cells affected are particularly metabolically active (and hence most sensitive to mitochondrial dysfunction). However, it is also possible that core mitochondrial functions in different neurons could be differentially dependent on different pathways and genes. As a result, the different genes affected in each disease might all regulate ETC function, but do so in a cell type–specific manner because the ETC itself is different in distinct cells. Future investigations of ETC function in different cell types will be needed to examine this notion.