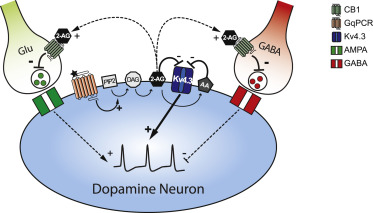
I'll wait for replication of their findings before getting excited. My guess is that it won't turn out to be important.
The finding just shows that calcium ion channels are affected in both ME/CFS and Long Covid patients. Calcium ion channels are regulated by cannabinoids.
Here is how cannabinoids are related to the symptomology of ME/CFS.
Leptin is raised in ME/CFS and upregulates a cannabinoid degrading enzyme called Fatty Acid Amide Hydrolase (FAAH) which degrades anandamide. Anandamide modulates various ion channels including T-type Ca2+ channels. Anandamide also helps modulate the HPA axis dampening its response and also modulates the immune system in the cental nervous system. Anadamide also modulates neuronal activity dampening it and modulating pain. It is also involved in stress resilience and affords a more resilient state of mind in adverse conditions and is thought to be suppressed in PTSD. It also regulates the gut.
Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide - PubMed (nih.gov)
Targeting Cannabinoid Signaling in the Immune System: “High”-ly Exciting Questions, Possibilities, and Challenges - PMC (nih.gov)
In the central nervous system (CNS), these processes include regulation of appetite, pain sensation, mood, and memory, whereas in the peripheral tissues, e.g., bone formation, spermatogenesis, sebum production, etc., and, maybe most importantly, immune functions (
7,
29–
37). Indeed, eCB signaling was shown to be an important orchestrator of both the innate and adaptive immune responses. Although there are some contradictions in the literature, ECS is generally considered to be a homeostatic “gate-keeper” of the immune system, preventing the onset of pathological, overwhelming proinflammatory responses.
Abstract
It is well known that certain active ingredients of the plants of
Cannabis genus, i.e., the “phytocannabinoids” [pCBs; e.g., (−)-
trans-Δ9-tetrahydrocannabinol (THC), (−)-cannabidiol, etc.] can influence a wide array of biological processes, and the human body is able to produce endogenous analogs of these substances [“endocannabinoids” (eCB), e.g., arachidonoylethanolamine (anandamide, AEA), 2-arachidonoylglycerol (2-AG), etc.]. These ligands, together with multiple receptors (e.g., CB1 and CB2 cannabinoid receptors, etc.), and a complex enzyme and transporter apparatus involved in the synthesis and degradation of the ligands constitute the endocannabinoid system (ECS), a recently emerging regulator of several physiological processes. The ECS is widely expressed in the human body, including several members of the innate and adaptive immune system, where eCBs, as well as several pCBs were shown to deeply influence immune functions thereby regulating inflammation, autoimmunity, antitumor, as well as antipathogen immune responses, etc. Based on this knowledge, many
in vitro and
in vivo studies aimed at exploiting the putative therapeutic potential of cannabinoid signaling in inflammation-accompanied diseases (e.g., multiple sclerosis) or in organ transplantation, and to dissect the complex immunological effects of medical and “recreational” marijuana consumption. Thus, the objective of the current article is (i) to summarize the most recent findings of the field; (ii) to highlight the putative therapeutic potential of targeting cannabinoid signaling; (iii) to identify open questions and key challenges; and (iv) to suggest promising future directions for cannabinoid-based drug development.
Daily cytokine fluctuations, driven by leptin, are associated with fatigue severity in chronic fatigue syndrome: evidence of inflammatory pathology | Journal of Translational Medicine | Full Text (biomedcentral.com)
Role for fatty acid amide hydrolase (FAAH) in the leptin-mediated effects on feeding and energy balance - PubMed (nih.gov)
Abstract
Endocannabinoid signaling regulates feeding and metabolic processes and has been linked to obesity development. Several hormonal signals, such as glucocorticoids and ghrelin, regulate feeding and metabolism by engaging the endocannabinoid system. Similarly, studies have suggested that leptin interacts with the endocannabinoid system, yet the mechanism and functional relevance of this interaction remain elusive. Therefore, we explored the interaction between leptin and endocannabinoid signaling with a focus on fatty acid amide hydrolase (FAAH), the primary degradative enzyme for the endocannabinoid
N-arachidonoylethanolamine (anandamide; AEA). Mice deficient in leptin exhibited elevated hypothalamic AEA levels and reductions in FAAH activity while leptin administration to WT mice reduced AEA content and increased FAAH activity. Following high fat diet exposure, mice developed resistance to the effects of leptin administration on hypothalamic AEA content and FAAH activity. At a functional level, pharmacological inhibition of FAAH was sufficient to prevent leptin-mediated effects on body weight and food intake. Using a novel knock-in mouse model recapitulating a common human polymorphism (FAAH C385A; rs324420), which reduces FAAH activity, we investigated whether human genetic variance in
FAAH affects leptin sensitivity. While WT (CC) mice were sensitive to leptin-induced reductions in food intake and body weight gain, low-expressing FAAH (AA) mice were unresponsive. These data demonstrate that FAAH activity is required for leptin's hypophagic effects and, at a translational level, suggest that a genetic variant in the FAAH gene contributes to differences in leptin sensitivity in human populations.
Protective effects of elevated anandamide on stress and fear-related behaviors: translational evidence from humans and mice - PubMed (nih.gov)
Abstract
Post-traumatic stress disorder (PTSD) is a common, debilitating condition with limited treatment options. Extinction of fear memories through prolonged exposure therapy, the primary evidence-based behavioral treatment for PTSD, has only partial efficacy. In mice, pharmacological inhibition of fatty acid amide hydrolase (FAAH) produces elevated levels of anandamide (AEA) and promotes fear extinction, suggesting that FAAH inhibitors may aid fear extinction-based treatments. A human FAAH 385C->A substitution encodes an FAAH enzyme with reduced catabolic efficacy. Individuals homozygous for the FAAH 385A allele may therefore offer a genetic model to evaluate the impact of elevations in AEA signaling in humans, helping to inform whether FAAH inhibitors have the potential to facilitate fear extinction therapy for PTSD. To overcome the challenge posed by low frequency of the AA genotype (appr. 5%), we prospectively genotyped 423 individuals to examine the balanced groups of CC, AC, and AA individuals (n = 25/group). Consistent with its loss-of-function nature, the A allele was dose dependently associated with elevated basal AEA levels, facilitated fear extinction, and enhanced the extinction recall. Moreover, the A-allele homozygotes were protected against stress-induced decreases in AEA and negative emotional consequences of stress. In a humanized mouse model, AA homozygous mice were similarly protected against stress-induced decreases in AEA, both in the periphery, and also in the amygdala and prefrontal cortex, brain structures critically involved in fear extinction and regulation of stress responses. Collectively, these data suggest that AEA signaling can temper aspects of the stress response and that FAAH inhibition may aid the treatment for stress-related psychiatric disorders, such as PTSD.
The role of the endocannabinoid system in the brain-gut axis - PMC (nih.gov)
Abstract
The actions of cannabis are mediated by receptors that are part of an endogenous cannabinoid system. The endocannabinoid system (ECS) consists of the naturally occurring ligands
N-arachidonoylethanolamine (anandamide) and 2-arachidonoylglycerol (2-AG), their biosynthetic and degradative enzymes, and the cannabinoid receptors CB1 and CB2. The ECS is a widely distributed transmitter system that controls gut functions peripherally and centrally. It is an important physiologic regulator of gastrointestinal motility. Polymorphisms in the gene encoding CB1 (
CNR1) have been associated with some forms of irritable bowel syndrome. The ECS is involved in the control of nausea and vomiting and visceral sensation. The homeostatic role of the ECS also extends to the control of intestinal inflammation. We review the mechanisms by which the ECS links stress and visceral pain. CB1 in sensory ganglia controls visceral sensation, and transcription of
CNR1 is modified through epigenetic processes under conditions of chronic stress. These processes might link stress with abdominal pain. The ECS is also involved centrally in the manifestation of stress, and endocannabinoid signaling reduces the activity of hypothalamic–pituitary–adrenal pathways via actions in specific brain regions—notably the prefrontal cortex, amygdala, and hypothalamus. Agents that modulate the ECS are in early stages of development for treatment of gastrointestinal diseases. Increasing our understanding of the ECS will greatly advance our knowledge of interactions between the brain and gut and could lead to new treatments for gastrointestinal disorders.
Amygdala FAAH and anandamide: mediating protection and recovery from stress - PMC (nih.gov)
Abstract
A long-standing literature linking endocannabinoids (ECBs) to stress, fear, and anxiety has led to growing interest in developing novel anxiolytics targeting the ECB system. Following rapid on-demand biosynthesis and degradation upon neuronal activation, the ECB
N-arachidonoylethanolamide (anandamide, AEA) is actively degraded by the serine hydrolase enzyme, fatty acid amide hydrolase (FAAH). Exposure to stress rapidly mobilizes FAAH to deplete the signaling pool of AEA and increase neuronal excitability in a key anxiety-mediating region – the basolateral amygdala (BLA). Gene deletion or pharmacological inhibition of FAAH prevents stress-induced reductions in AEA and associated increases in BLA dendritic hypertrophy and anxiety-like behavior. Additionally, inhibition of FAAH facilitates long-term fear extinction and rescues deficient fear extinction in rodent models by enhancing AEA–CB1 (cannabinoid type 1) receptor signaling and synaptic plasticity in the BLA. These preclinical findings propose restoring deficient BLA AEA levels by pharmacologically inhibiting FAAH as a mechanism to therapeutically mitigate the effects of traumatic stress.
Endocannabinoid Signaling, Glucocorticoid-Mediated Negative Feedback and Regulation of the HPA Axis - PMC (nih.gov)
Therefore, the role of the endocannabinoid system in HPA axis regulation extends beyond the acute stress phase to chronic stress plasticity, and the endocannabinoid system plays an integral part in the changing expression of the HPA response to a continually fluctuating and often challenging environment.
Abstract
The hypothalamic-pituitary-adrenal (HPA) axis regulates the outflow of glucocorticoid hormones under basal conditions and in response to stress. Within the last decade, a large body of evidence has mounted indicating that the endocannabinoid system is involved in the central regulation of the stress response; however, the specific role endocannabinoid signalling plays in phases of HPA axis regulation, or the neural sites of action mediating this regulation, was not mapped out until recently. This review aims to collapse the current state of knowledge regarding the role of the endocannabinoid system in the regulation of the HPA axis to put together a working model of how and where endocannabinoids act within the brain to regulate outflow of the HPA axis. Specifically, we discuss the role of the endocannabinoid system in the regulation of the HPA axis under basal conditions, activation in response to acute stress and glucocorticoid-mediated negative feedback. Interestingly, there appears to be some anatomical specificity to the role of the endocannabinoid system in each phase of HPA axis regulation, as well as distinct roles of both anandamide and 2-arachidonoylglycerol in these phases. Ultimately, the current level of information indicates that endocannabinoid signalling acts to suppress HPA axis activity through concerted actions within the prefrontal cortex, amygdala and hypothalamus.
Endocannabinoid system, stress and HPA axis - ScienceDirect
Abstract
The
endocannabinoid system (ECS), which is composed of the
cannabinoid receptors types 1 and 2 (CB1 and CB2) for marijuana's psychoactive ingredient ∆9-tetrahydrocannabinol (∆9-THC), the endogenous ligands (AEA and 2-AG) and the enzymatic systems involved in their biosynthesis and degradation, recently emerged as important modulator of emotional and non-emotional behaviors. In addition to its recreational actions, some of the earliest reports regarding the effects of
Cannabis use on humans were related to
endocrine system changes. Accordingly, the ∆9-THC and later on, the ECS signalling have long been known to regulate the hypothalamic-pituitary-adrenocortical (HPA) axis, which is the major neuroendocrine stress response system of mammals. However, how the ECS could modify the stress hormone secretion is not fully understood. Thus, the present article reviews current available knowledge on the role of the ECS signalling as important mediator of interaction between HPA axis activity and stressful conditions, which, in turn could be involved in the development of
psychiatric disorders.
Am I making my point yet
")