Questions for Prusty (paper summarised
above).
Questions asked
and replied to on this tweet. Prusty's responses in quotes:
(1) Does time frame of transfer effect suggest mechanism for patient's 1-2 day delayed PEM, crashes, etc? Naviaux's previously said [
2018] that "Mitochondria change their function rapidly under stress. Within minutes[...]". But you cultured the HHV-6 transactivated cells for 2 days and then the responder cells for 2 days in the transferred supernatant.
(a) Were these durations necessary for the phenotype transfer to work? (I.e. to accumulate or respond to the molecular factor.) Did you try shorter times to find a minimum?
(b) Or was the duration part of technical requirements of the analysis methods?
Ql: PEM related crashes and fatigue is a complex physiological process and can involve many factors. We are in the process of understanding this complex phenomenon. Mitochondrial fragmentation is a very quick process under experimental conditions. With a sufficient stimulus, one can induce mitochondrial fragmentations within 2-3 h. But this is not how it works in nature. The CDR response is initiated from a limited number of cells and it requires sufficient time to reach every cell and completely shut down every cell. This is usually in the range of 2-4 days depending upon the number of original events. Yes, we can see effects starting to show up within 24 h. But the statistically significant effect that we want to show takes a longer time. In short, the metabolically and energetically crunch time is the hardest time that the cell or the body faces, when every other alternative is exhausted. [
Twitter]
(2) Did this paper show cells in a CDR1 state? "M1" mitochondria and anti-viral protection are characteristic of Naviaux's CDR1 state. But he categorises ME/CFS as being a CDR3 disease [
2019, Fig.2]:

(a) Was he possibly mistaken about ME/CFS being CDR3 associated?
(b) Or are the majority of our cells stuck in CDR3 as a result of a smaller population of cells in CDR1 (e.g. with HHV-6 transactivation) preventing completion of the healing cycle?
Q2: This is what I also asked Bob when I had my results and he explained me very nicely, which is not possible to do it here. Just to tell you that M1 mitochondria is the shape of mitochondria when they are under stress like PEM. But if the mitochondria remain in M1 form forever, the cells will simply die (mitophagy). This possibly happens in neurons causing neuronal damage in long run. The M2 form is probably preferred under resting conditions. But unfortunately, we cannot test this in the body. [
Twitter]
(c) Or are the your observations not sufficient to draw conclusions on this because they are in cancer cells?
These cells are treated with both control & patient serum. So cell line does not make any difference here. [
Twitter]
Q2: Our work is done in cancer cell lines as it is necessary for creating the HHV-6 reactivation system. All the other work are in cancer cell lines because we need to have a uniform system to work. [
Twitter]
(d) Are the lab cancer cells naturally in a proliferative CDR2 state...?
(3) Was
Naviaux's contribution to the paper purely advice and/or interpretation (no lab work)?
Q3: Bob has helped in me in making a lot of sense out of my work. He has not done any actual experiment for this paper. But we are continuing on some of the cool stuffs now [
Twitter]
(4) Which cells in patients are (or aren't) affected?:
(a) Are initial HHV-6A infections mostly limited to cells with higher CD46 expression (chart below)? And HHV-6B to cells with CD134, primarily CD4 T-Cells, etc?

Q4: CD46 is expressed in every nucleated human cell. Hence HHV-6 can infect virtually every nucleated human cell. Same is also true for HHV-6B. T-cells only allow productive virus infection, which is clinically not that relevant in my personal opinion. [
Twitter]
(b) Was HHV-6A virus chosen, instead of 6B or 7, too be able to infect U2-OS and A549
We wanted to have a unique fluorophore-based system for our study. As HHV-6A is the only virus out of the three (HHV-6A, HHV-6B and HHV-7), whose genome is available in the form of a BAC, we chose this one for our work. We will continue working with the rest of the two in future. The cell type in which the virus is integrated and where (chromosomal site) it is integrated, might influence the rate of reactivation and hence the severity of the disease. [
Twitter]
(c) Could the extent of the initial (latent) infection determine the severity of ME/CFS, after the reactivation triggering event has passed?
(d) Or is infection and reactivation in specific locations more likely to be key? E.g. You've found infections all over the brain and brainstem [
YouTube].
(e) Could worsening of ME/CFS severity come from a spread of HHV-6 infection to more cells?
I do not think that these viral reactivation spreads from cell to cell as there is literally no virus particle formation. Virus particle formation would be the simplest infection scenario possible as the host immune system would then recognize it and will eliminate it. [
Twitter]
(5) Why do most people *not* get ME/CFS after acute infections, surgery, etc? If everyone has HHV-6 (and other viruses) latent in their bodies. (Some factors in 3, genetic and/or metabolic status?)
Q5: People do reactivate these viruses from time to time. But most of the times, our own immune system takes care of it. We do not even realize it. But in some of us, there must be a genetic component that makes us susceptible to conditions like ME/CFS. We are trying to understand that. [
Twitter]
(6) How does HHV-6 transactivation become chronic in patients?
(a) The U2-OS and A549 cells are incapable of late stage viral replication. Is this also true for some cells in the human body (that can be infected)?
(b) Does the virus deliberately prevent itself from complete replication? What's the evolutionary advantage, if so? Is it related to blocking competing viruses from getting its host cell destroy by the immune system...?
(c) Is another mechanism needed for chronic HHV-6 activation? e.g. another infection, accumulation of toxic elements or other researcher's hypothesis (below)?
Q6: In our body, most of the cell types does not allow productive HHV-6 infection. Only CD4+ cells allow productive HHV-6 infection. HHV-6 would never want to get fully eliminated from the cell. So, it would prefer to stop itself being detected as soon as it starts reactivating. The Immediate early (IE) viral RNA would immediately be detected in cell cytoplasm by host innate immune machinery. Hence it starts producing small non-coding RNAs first, which helps in preparing the stage for further virus reactivation. It is a constant battle between the cells and the virus. Sometimes the cell wins and sometimes the virus.
An ideal cellular environment is the most important requirement for fully productive virus infection. [
Twitter]
(7) Do you suspect interactions with any other proposed mechanisms? I.e. as them causing sustained HHV-6 transactivation, or vice versa? Specifically:
(c) Michael VanElzakker's "vagus nerve infection" hypothesis [
2013]?
(d) Any others you have an eye on...? (E.g. research on ROS/oxidative stress.)
(8) Any hints of patient symptom phenotypes being separable by something you've measured...?:
(a) Detectable U14 (in 40% of sampled patients)?
(b) Inherited ciHHV-6?
(c) What about the never sick patients verses those who catch everything (or at least have repeatedly infection symptops).
(9) ATP production:
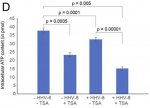
(a) Was this shown halved in the U2-OS cells, upon transactivation or with transfered supernatant. But a much more marginal reduction in the A549 cells (am I'm reading the paper right)?
(b) Does [
Fig.2D] show halved ATP production purely due to application of TSA? (If so, is that a big distorting factor on results?)

Q9: The figure 2D is the ATP content in U2-OS cells after adoptive transfer of the culture supernatant. The results are more or less the same in A549 cells. TSA do have some effect, but it is very clear that virus reactivation itself has a stronger effect. If one normalizes the data to the TSA added sample, the difference still stands clear and significant.
The A549 ATP assay that you see in figure 5 is from patient serum treated cells. I guess we need to grow the cells for longer time to see a more significant effect on cellular ATP content. But I do not think that we can see a dramatic decrease in ATP content under any condition. Minor changes in Physiological levels of ATP can have a drastic effect on cells. [
Twitter]
(c) Are the measured ATP concentrations indicate a matching scaling of flux (in production rate)?
No, we did not measure the production rate of ATP. [
Twitter]
(d) Would you expect to see a similar contrast in effect between different tissues within a patient? Or between patients? Or are these results not indicative at all, because they are cancer cells?
Again, cancer cells have a different physiology. But until we have another system, we have to depend upon these cells for our work. In future, we will try utilizing primary cells for such work.
Of course, different tissues or cells will show different levels of effect. [
Twitter]
(e) Why is there a factor of 10 difference between ATP concentrations graphed in fig.2d verses fig.1c...? (An error?)
Good observation. This depends upon the number of cells being used for the study. We did not show ATP content per cell. We take similar number of cells per experiment. The figure 1C was from experiments done a long time back. Figure 2D was done during the revision. As we have used more cells for 2D, we have almost 10 times more ATP content. [
Twitter]
(10) Issue with the paper - Does the last sentence of your abstract seems to claim too much? You clearly showed strong similarities but no direct evidence of causation by HHV-6 in patients; it was an in vitro study. Is there unpublished work that bridges this gap?
Q10: I do not think that it will ever be possible to show HHV-6 infection in ME/CFS patients causing the disease. It is not a localized disease like cancer, and it is not an overnight disease. So, it is up to you to decide whether we claim too much or not. We do not claim that we solved the mystery. [
Twitter]
(11) Serum factor...:
(a) Can you give us any more hints about what molecules you are currently honing in on, or the nature of the tests you've mentioned?
Q11: We are trying everything that we can think about. I am not in a position to tell you anything. It is a very competitive field. If I tell you anything, someone will come out and tell that it is too early, and we do not have enough data to support. So let us work and come to stronger results. [
Twitter]
(b) Could the U14 protein be the factor? Seeing as it turns up in so many pathogens. But you only found it in 40% patient serum, so would that make it one of multiple factors? Or could it be pressent in all, but below your detection threshold?
No, I do not think that U14 protein is a factor. I guess you are talking about U14 small non-coding RNA. It is very much possible that other viruses have similar RNA. But our probes are extremely stringent and do not detect anything having more than one nucleotide difference. So, we are definitely not detecting any similar RNA from other herpesviruses. It needs tons of money to test all the other herpesviruses. We will do it systematically as and when possible. [
Twitter]
(c) Have you been able to rule out any of the types of molecule you mentioned previously [
YouTube]? I.e. Mitochondrial metabolites, exosomes containing small non-coding RNAs or proteins, cellular RNA, antibodies, calcium flux...? And what about purinergic signalling (ATP, etc)?
Yes, we are ruling out some of them. But once again, I cannot tell anything about it at this moment. [
Twitter]
(a) Gradual onset - can this work on latent virus reactivation fit with (very) gradual onset of ME/CFS?
Q12: Yes, I think so. Virus reactivation and its consequential effects are slow process. It matches well with the slow onset process of the disease. [
Twitter]
(b) Predisposing factors - could deleterious SNPs of SOD2 (or methylation enzymes, upregulated COMT, etc) make viral activation more likely? Or its effects more pronounced?
Yes, it can have consequences. [
Twitter]
(c) Delayed sleep - Could the CDR state (or viral proteins) directly slow down the 24h clock gene expression pattern? Specifically, the
astrocytes in the SCN seem to be the body's master time keepers. Because circadian rhythm is so commonly delayed in the illness - probably more of a high level neurological issue (but it was my first symptom, worsening with very gradual onset).
The link to circadian clock is fascinating. Some of the ME/CFS patients have spoken to me on this. I am very much fascinated with the idea and the literature do support an association between circadian rhythm and mitochondrial morphology. I think hormonal changes, metabolism and other factor that has a link to circadian rhythm can make the situation more interestink. I would love to test this in future if time and finances permits. [
Twitter]
:
(a) Can HHV-6 DNA show up sometimes in whole genome sequencing? I assume this is deliberately filtered out of the final data, even if its chromosomally integrated...?
Yes, one can see HHV-6 in whole genome data. But an extremely high sequencing depth is required to see these sequences. Yes, most of the times these sequences are discarded during initial data filtration. [
Twitter]
(b) How close are we to having a working CRISPR system that could remove HHV-6 from humans (in vivo)? Would have to do this by providing immunity, blacklisting the key viral proteins, or something?
Some of my colleagues are working on CRISPR technology to remove integrated HHV-6 from the genome. But I guess we are still very far away from any successful results. [
Twitter]
(c) Are there any exciting new laboratory technologies or equipment you expect to have access to in the near future?
We keep developing exciting technologies and also keep looking for new ones developed by others. It is hard to say here what we really want. [
Twitter]


") More so that they were able to make this signal (or one with identical effects) in a modified standard laboratory cell line. Able to turn the signal on and off on demand.
More so that they were able to make this signal (or one with identical effects) in a modified standard laboratory cell line. Able to turn the signal on and off on demand.