

necessary8
Senior Member
- Messages
- 134
The RBC-NOX hypothesis
Hello again, everyone. Welcome, to the RBC-NOX hypothesis, which I consider by far my best idea yet.
It connects the IDO metabolic trap, red blood cell deformability, craniocervical instability (including recoveries after surgery), Alan Light’s research on fatigue signaling, lactate, extracellular ATP, parts of the immune system, mold exposure, minor factors like selenium and B12, and explains almost all of Ron’s nanoneedle data, including plasma switch results and SS-31, as well as possibly Prusty’s mitochondrial fission results. Symptom-wise, it explains fatigue, brain fog, digestive symptoms, and possibly also sensory sensitivity, cognitive problems, sleep problems and pain. Most of all, it provides an especially good explanation for PEM with all of it’s weird features. It accounts for viral onsets as well as gradual and other atypical onsets.
Erythrocyte deformability and fatigue signaling
To start, we must first talk about erythrocyte (red blood cell) deformability. As most of you know, Ron’s group observed it to be clearly impaired in ME/CFS patients, and there have been studies showing similar data before.
During even minor muscle contraction, the local need for oxygen goes way up, and so there needs to be increased perfusion to accommodate that. If RBCs can't deform properly as they pass through the very narrow capillaries, trying to supply the muscle with extra oxygen, what will happen? Increased shear stress on the vasculature, and possible very local hypoxia. Both of those dependent very closely on muscle activity, as during rest there is probably enough oxygen for the cells to function normally. This means that the muscles will much faster switch to anaerobic energy production, than in healthy people. Meaning more lactate in small exercise, but normal at rest.
Now, the issue of lactate in ME/CFS has been a bit of a contentious topic, I’ve seen a lot of people making very different claims about it, and there hasn’t been many good studies that examined it up close. This changed recently, with this publication: https://physoc.onlinelibrary.wiley.com/doi/10.14814/phy2.14138
Although the sample size is only 18, they used rigorous canadian criteria and their p values are very good. Their findings are that lactate in ME/CFS is normal at rest, but abnormally high after small exercise. The same thing was found by Mark Vink, a researcher who got sick with ME/CFS and did the observations on himself: https://www.researchgate.net/public...gic_EncephalomyelitisChronic_Fatigue_Syndrome
It is also consistent with Naviuax’s metabolomics data, in which lactate is normal (because the measurement was made at rest only).
For me that is a pretty good consensus in the data. Normal at rest, very high in small exertion. This fits with what I proposed earlier - that it is a consequence of impaired RBC deformability.
So that is one consequence of it. What are the others? Well, the body will try to compensate for it. Sensing hypoxia and increased shear stress on the vasculature, the vascular cells as well as erythrocytes themselves will release two things: nitric oxide (NO), and extracellular ATP.
https://www.researchgate.net/profil...d1/Nitric-oxide-erythrocytes-and-exercise.pdf
https://pdfs.semanticscholar.org/85b4/76f74c3d80375cf9012cf1c9505e8ff0e6bc.pdf
https://www.researchgate.net/profil..._adenosine/links/0a85e5391794cde68e000000.pdf
The ATP part is hard to verify, because it gets degraded very quickly so you can’t measure it from the blood reliably. But NO you can, and sure enough, there is one study, with a decent sample size, showing higher NO levels in ME/CFS patients even at baseline, and much higher spike in NO in response to exercise, than in healthy people: https://www.liebertpub.com/doi/abs/10.1089/jwh.2008.1255
I will come back to nitric oxide later, but for now let’s consider ATP and lactate. Alan Light has done extensive research on how fatigue works in general, how does it happen. He had a talk about it in the stanford symposium, and I’ve also talked to him quite a bit about it. His conclusion is that lactic acid (so lactate + protons) and ATP have to both be released to the extracellular space surrounding the muscle, for the nearby nociceptive neurons to sense it and send the fatigue signal to the brain. And that does indeed happen normally in strenuous exercise, and it can be even artificially induced. Here is his paper about it: https://physoc.onlinelibrary.wiley.com/doi/full/10.1113/expphysiol.2013.075812@10.1002/(ISSN)1469-445X(CAT)VirtualIssues(VI)bbep2016
And so with this knowledge we can now see the first direct way in which decreased RBC deformability causes ME/CFS symptoms - because it causes ATP and lactic acid to be released much more easily from muscle activity than in healthy people, it will mean that the sensory fatigue neurons will perceive very light muscle exertion as strenuous exercise, and send signals to the brain accordingly. It can also explain brain fog and mental fatigability, because the brain has its own similar fatigue system that also senses extracellular ATP and lactate. And if we’re using our brain more than baseline, the brain also needs more oxygen, and runs into the same problem of impaired erythrocyte deformability, triggering the fatigue system much easier than in a healthy person, resulting in the feeling of mental fog, full body fatigue, and sometimes headaches.
One possible problem with this explanation is the fact that this accounts for activity-induced fatigue only (albeit with much more sensitivity to activity). And yet patients still have fatigue at rest. Perhaps I can explain it like this: patients who are mild still have a lot of physical activity so the fact that lactate+ATP are produced easier in them, is enough to make them fatigued all the time. And severe people still have some mental activity, which could generate lactate+ATP enough to activate nearby fatigue sensors in the brain itself, but not enough to be detectable in the bloodstream. And since activation of the fatigue system of the brain feels like full body fatigue, that could account for their fatigue at rest.
But perhaps there is more to it, I’m not sure.
The Why
So why is erythrocyte deformability impaired in the first place? This is where it gets interesting.
RBC deformability is naturally impaired by oxidative damage. This damage is accumulated over the life of the erythrocyte, which gradually loses its deformability, until being removed from the circulation. A lot of this is due to hemoglobin autoxidation to methemoglobin, and oxidative damage to band 3. This is an amazing paper about this process: https://www.frontiersin.org/articles/10.3389/fphys.2014.00084/full
And sure enough, there has been research showing increased oxidative damage in ME/CFS erythrocytes, including increased methemoglobin: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.505.9883&rep=rep1&type=pdf
They also observed increased levels of stomatocytes, which is a classification of erythrocyte shape. There isn't really a lot of research about stomatocytes, but there is a condition called hereditary stomatocytosis, and I found this old case study: http://www.bloodjournal.org/content/bloodjournal/46/5/659.full.pdf?sso-checked=true where stomatocytosis in a patient was found to be caused by increased permability of erythrocytes to calcium, with which the Ca-ATPase couldn't keep up, to pump it out.
This matches up with the big Mohanty paper I linked above, where they also state that oxidative damage to the Ca-ATPase is the main mechanism by which oxidative stress impairs erythrocyte deformability.
So what could be causing this increased damage in ME/CFS? Well, the fact that an ion pump is involved kind of rang a bell with me regarding Ron’s nanoneedle experiments. What if the same factor could be damaging erythrocyte Ca-ATPases (causing deformability issues), and Na/K-ATPases in white blood cells (causing the impedance signal in response to salt stress)?
And since he recently pointed to exosomes as responsible for the impedance signal, I searched if I could find something that would fit this. And I found something very interesting.
There are two demonstrated cases of exosomes containing functional NOX enzymes (NADPH oxidase), and producing ROS (reactive oxygen species) by themselves. One paper shows the production of them from platelets in sepsis: https://journals.lww.com/ccmjournal/Abstract/2004/03000/Platelet_derived_exosomes_of_septic_individuals.32.aspx
And the other, by infiltrating immune cells in neuronal damage: https://www.researchgate.net/profile/Francesco_De_Virgiliis/publication/323127843_Reactive_oxygen_species_regulate_axonal_regeneration_through_the_release_of_exosomal_NADPH_oxidase_2_complexes_into_injured_axons/links/5a8ac40b0f7e9b1a9554b80d/Reactive-oxygen-species-regulate-axonal-regeneration-through-the-release-of-exosomal-NADPH-oxidase-2-complexes-into-injured-axons.pdf
This fits perfectly, because if the source of ROS is outside of the cells, that means the proteins most susceptible to the damage will be the membrane-bound ones. And ATPases are one of the most abundant membrane-bound proteins.
Furthermore, the Mohanty paper on erythrocytes also states that they are very resistant to intracellular ROS because of a robust anti-oxidant system. But exogenous ROS are much more problematic for them.
This also explains why the nanoneedle signal is abolished by SS-31, which is a ROS scavenger.
There is even some research showing that ROS might mediate mitochondrial fission ( http://diabetes.diabetesjournals.org/content/diabetes/62/11/3851.full.pdf ), which could explain Prusty's findings (presented at the NIH conference).
And that is the core of this hypothesis. I propose that the exosomes in ME/CFS contain functional NOX enzymes, and are hence responsible for both the RBC deformability and the nanoneedle results. And the RBC deformability is what causes a lot of fatigue and makes patients very sensitive to physical and mental exertion.
Connection to the IDO metabolic trap
As many of you know Robert Phair’s IDO metabolic trap hypothesis is gaining more and more strength as the genetic testing reveals more and more ME/CFS patients with damaging IDO2 mutations, and not a single one without. To the point where it is increasingly probable that the IDO trap does happen *somewhere* in ME/CFS patients.
The hypothesis that I’m presenting here does not necessarily require the IDO trap to be taking place. But it does get more interesting if it does. Because it allows for a pretty good model of PEM.
Now, to the best of my knowledge the main version of the IDO trap that Rob and Ron are currently pursuing is that it would happen in serotonergic neurons of the raphe nuclei in the midbrain, where it would mediate most of the symptoms, including fatigue and PEM. It could also happen in immune cells accounting for some of the quasi-autoimmune observations, and independently in the gut, mediating the digestive symptoms. But everything else, all other symptoms would be contained in the midbrain.
I personally do not believe that is very probable. I can’t definitively prove that it isn’t happening, but when I look at the lactate studies I mentioned at the beginning, or at the plasma switch results either from Ron or Prusty, that is speaking to me that the disease mechanism happens, at least partially, in the periphery. That data just doesn’t make much sense if it would be contained in the brain.
And so, I propose a different main role for the IDO trap.
If you remember one of the papers on NOX-containing exosome production showed that immune cells can do it, in specific conditions. Now, this is a bit of speculation on my part, but perhaps there is a limiting mechanism of such exosome production, that relies on IDO activity. It is not a far fetched idea, considering that negatively regulating other immune cells is the main way in which the immune system uses IDO. Eg: https://science.sciencemag.org/content/297/5588/1867
So let’s imagine a person who has a damaging mutation to IDO2. That person, through a viral infection or another trigger, gets some of their immune cells stuck in the IDO metabolic trap. At the same time, something else happens which requires the immune system to make some NOX-containing exosomes. But the limiting mechanism for this exosome production is disabled, because of the IDO trap. So it makes more of them than it needs to, and they are not as contained locally as they should. They seep into the bloodstream, damaging cell surface ATPases of all cells in the blood. Including erythrocytes. Which now are less deformable, and cannot keep up with the oxygen demand in physical or mental exertion. Producing too much eATP and lactate, causing all the fatigue. And producing also more nitric oxide. I promised I’m gonna go back to the nitric oxide, right?
Well, you see, NO actually inhibits IDO activity: http://www.jbc.org/content/269/20/14457.full.pdf
A year ago when I talked to Rob Phair about PEM, he suggested that nitric oxide could be the factor that ties IDO to exercise. And I subsequently showed him that the levels of NO produced in normal exercise are much lower than the ones needed to inhibit IDO. So it doesn’t work.
But! With RBC deformability issues, that changes. As I mentioned before, it makes it so that much more NO is produced much easier. And I’m gonna again link the paper showing that indeed this is happening in ME/CFS patiens: https://www.liebertpub.com/doi/abs/10.1089/jwh.2008.1255
If the change is that big in the blood, in the tissues themselves the NO levels will probably be high enough to actually inhibit IDO, and make the trap in some of the immune cells even worse, which will make them even worse limiters of exosome production, resulting in more exosomes, and more oxidative damage to erythrocytes. Basically making the whole disease worse. But only when a patient crosses a certain threshold of exertion, which would generate enough NO to do this. Sound familiar? Yes, this is how I propose PEM would work. It can roughly account for the time delay, I think. It can explain why PEM can be caused by both mental and physical exertion, and why during recovery from one PEM episode, it is much easier to cause another one.
It can also explain what the deal is with digestive symptoms. I’m sure many of you have noticed, but digestive symptoms in ME/CFS are pretty indistinguishable from IBS. But for most patients, they came about together. Sometimes maybe not right away, but following them getting sick with ME/CFS. But also, there are a lot of people who have IBS and don’t have ME/CFS. And the mechanism of IBS is also largely unknown.
So perhaps, IBS is just the IDO trap taking place in gut cells. You can have it without ME/CFS, without the whole RBC deformability, NOX exsosomes, etc. You can get it in some way as long as you have the IDO2 mutation.
But when you do get ME/CFS, because of the increased nitric oxide during crashes, sooner or later in most people it pushes their gut cells into the IDO trap as well, resulting in the digestive symptoms.
In a bit of a similar vein we could perhaps explain the relationship with autoimmunity. I don’t remember who did this study, but someone at the Stanford symposium presented data showing that the families of ME/CFS patients have very high incidence of autoimmune disease. And there has long been talk of whether ME/CFS is autoimmune or not. But despite a lot of efforts we still do not have a clear autoantibody or a self target, and rituximab ultimately failed. And yet the familial correlation remains. So here is a crazy thought - perhaps a lot of autoimmune diseases are caused by the IDO trap happening in the bone marrow? To me it seems very possible, because IDO activity is used in the bone marrow to directly inhibit auto-reactive T cells: http://www.bloodjournal.org/content/bloodjournal/103/12/4619.full.pdf?sso-checked=true
And so, the IDO2 damaging mutations, being hereditary, would predispose to both autoimmune diseases as well as ME/CFS. Hence the familial correlation, without ME/CFS being strictly autoimmune.
This doesn’t really have much to do with the rest of what I’ve written here, but I wanted to mention it, cause it’s interesting to think about.
Connection to craniocervical instability
As you may know, recently Jen Brea have recovered from her ME/CFS following spinal fusion surgery to correct her craniocervical instability (CCI). This brought to light earlier stories of other patients who had a similar experience, including @jeff_w who described his story in a very well put together blog (https://www.mechanicalbasis.org/ ) I have been following this angle for quite some time, even before Jen went through her surgery. I believe that we can learn a lot about ME/CFS by looking at those cases, even if they end up being a small, small minority.
My immediate reaction was that we need to test those patients with nanoneedle before and after surgery, and I’m hearing that Ron’s group intends to do that. We will know much more once they do.
But in the meantime, I can offer you some possible explanations for how this might work.
When I first heard Jeff’s story, the most probable explanation to me seemed that perhaps the mechanical compression of the midbrain (which happens in CCI) can produce the exact same symptoms as the IDO trap taking place in the midbrain (which, as I mentioned, was the hypothesis of Rob Phair)
I still think that option is possible, but as I read more, I started having doubts.
The RBC-NOX hypothesis offers another explanation, perhaps a more probable one. If you remember, one of the two documented cases in which the body produces NOX-containing exosomes, was to mediate neuronal repair. Perhaps, because of the damage to the neurons being compressed in CCI, the immune system reacts by trying to repair as much as it can, generating NOX exosomes for it. And because the damage is ongoing, it never stops. At the same time, the limiter for this is disabled because of the IDO trap, allowing a lot of those exosomes to be made, escaping to the bloodstream and causing damage to cell surface ATP-ases, resulting in ME/CFS symptoms, and tying them very closely to physical exertion. And then, when CCI is fixed, and the body recovers, there is no need for the immune system to produce the exosomes. And so it stops. And ME/CFS goes away.
Perhaps in other groups of patients the reasons for NOX exosome production could be different things. It doesn’t always have to be CCI, it doesn’t even have to be neuronal damage. I’m convinced those exosomes are probably used for other things too, the subject just hasn’t been studied very closely. There are very few things in the human body that do only one thing.
Connection to toxic mold
One of those other things might actually be mold exposure. As many of you know there is a significant subset of patients whose illness closely depends on exposure to molds. If they’re around mold, they get worse, if they are away from it, they get better. It’s not all patients, not even a majority probably, but it’s a significant enough portion that you can’t chalk it up to coincidence. And I haven’t really seen many hypotheses which would explain why that is the case, and which would make sense with other research findings. So here is my take:
It’s actually pretty widely documented that both mold-originating bacteria, as well as many mycotoxins induce NO and ROS production (sometimes through NOX) in human cells:
https://www.sciencedirect.com/science/article/pii/S0300483X97001418
https://www.sciencedirect.com/science/article/pii/S0300483X1200337X
https://www.sciencedirect.com/science/article/pii/S0887233309001842
Now, the research mostly focuses on ROS as the means of damage to the cells, and it does not mention exosomes. But it is not far-fetched to speculate that perhaps immune cells would release NOX-containing, ROS-producing exosomes if not to fight with the pathogens, instead to signal their presence to the rest of the immune system. This makes sense from an evolutionary standpoint, because if toxic mold exposure always meant ROS in the system, then organisms could develop ways to release ROS themselves to signal the presence of this pathogen.
And hence mold exposure in this subset of patients could play the same role as CCI in others - making the immune system produce NOX exosomes, which in combination with the disabling of the immune limiter of this process (by the IDO trap), results in ME/CFS.
(I will admit I haven’t looked into this angle as close as I should, because it is a rabbit hole of its own. I might be very wrong here)
Visual diagram and summary
If you’re feeling a bit lost at this point due to the complexity of this hypothesis, here is a diagram that should help:
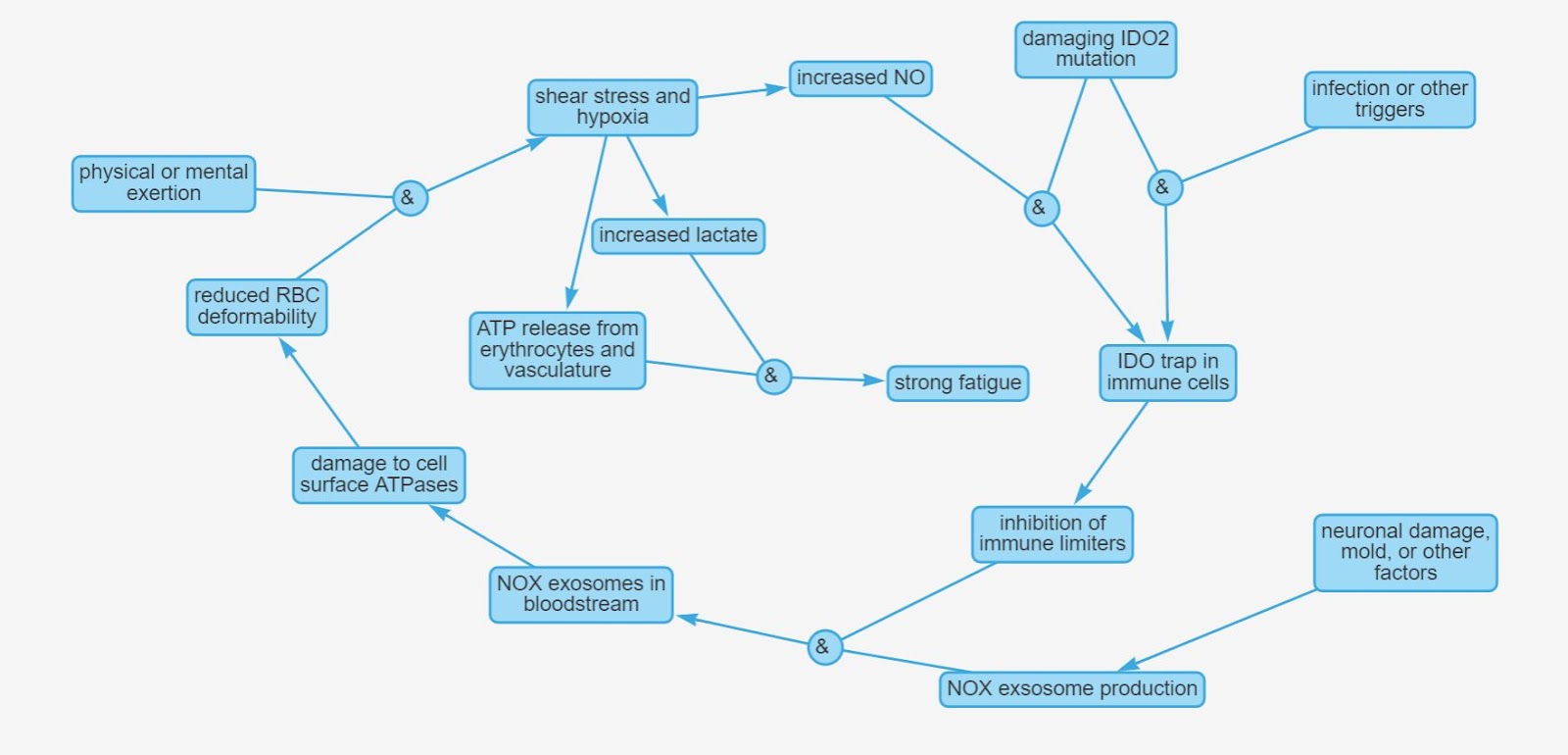
Basically, I propose that ME/CFS is the result of three factors that all need to coexist for the illness to take place:
1. Genetic mutation in IDO2
2. A trigger that will push key immune cells into the IDO trap, like an infection or something else - this only needs to happen once, after that the cells stay in the trap by itself
3. A factor that would make the immune system produce NOX-containing exosomes, like neuronal damage, mold exposure, etc - this one needs to be ongoing
Other symptoms
There are a lot of other symptoms that are common in ME/CFS which I would like to account for in this model, but since I don’t feel like I have the full story on them yet, I will only mention them briefly.
Pain uses a lot of the same pathways as fatigue, so a lot of the muscle pains people have can be explained in the same way as fatigue.
Nitric oxide plays some role in regulating sleep (https://www.sciencedirect.com/science/article/abs/pii/S1087079204000681 ), although it’s not very well understood yet. So perhaps it is possible that the high NO levels in ME/CFS lead to some desensitizations of the pathways involved with sleep, and they don’t respond properly to its normal NO modulation, causing sleep problems.
Neurons also happen to have cell-surface ATPases which are pretty vital to their function. So if we have circulating NOX-containing exosomes, they could as well damage some of those ATPases, which could possibly explain sensory oversensitivity, cognitive issues, memory problems and pain. But for this to be a really solid idea, I would need to explain why those neural pathways in particular are more sensitive to that type of damage than other ones. Which I can’t yet.
Other possible small connections
Since selenoproteins are antioxidants that can also act outside of the cells ( http://www.jbc.org/content/283/29/20181.full.pdf ), this can explain why low selenium can be a small cofactor (another one of Ron’s findings), and why some patients found small long-term improvement with high doses of selenium. But it also makes sense that it wouldn’t cure people.
Same thing with B12.
A phenomenon that is known to a lot of patients and has gone largely unexplained is the fact that some pregnant patients get remissions during pregnancy. An interesting possibility to consider here is the fact that in pregnant women, there is a 30% increase in 2,3-BPG, which shifts the oxygen dissociation curve, allowing for easier liberation of oxygen from hemoglobin. This could perhaps, in some individuals, overcome the RBC deformability issues, and supply enough oxygen to exercising muscle and active brain regions, abolishing a lot of the symptoms.
A lot of the immune dysfunctions observed in ME/CFS could possibly be explained by either the IDO trap happening in immune cells, or by the oxidative damage to cell surface ATPases. Maybe. I don’t have any rigorous analysis to support this notion, just the general idea that it should be possible.
Moving forward
The main prediction that this hypothesis makes is that the exosomes present in ME/CFS plasma will contain functional NOX enzymes. If that is not the case, most of it falls apart.
Now, I am very, very far from being knowledgeable about the technicalities of experimental design, but if my shallow understanding is correct, then this prediction should be fairly easy to test. As long as you can do exosome separation, it should suffice to just western blot the exosomes for all known NOX isoforms, and compare data on that from patients to controls.
I have sent this to Ron’s team already (well, not the entirety of this post, but the bare core of the hypothesis was there), about 3 months ago, but since I still haven't gotten any word back, they either are so swamped with other stuff that they didn’t read it, or they know something that I don’t, something that makes this hypothesis bad, and they didn't wanna waste time telling me about it. Both are possible, though I'm leaning towards the former.
So I figured I’d post it here. If I made logical mistakes or false assumptions, hopefully you guys can point them out to me. And If I didn’t, perhaps you can help me get a different research group involved to test this, if Ron doesn’t want to (which I still don't even know if he's seen it or not).
Here is a Google Doc version if you need to send it somewhere: https://docs.google.com/document/d/1nqFiBMqz76VmyJ2yVVfFj7W-7L5Ra4q9Ym-lFgmGEEU/edit?usp=sharing
Tagging also @Hip @nandixon @Murph cause they know a lot
If anyone has questions, I will do my best to answer them.
Last edited: