Hip
Senior Member
- Messages
- 18,313
@nandixon
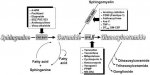
More on ketamine as a mTOR activator: this study says ketamine induces mTOR activation:
They also mention that:
However, this idea that boosting mTOR may be helpful in ME/CFS does not fit in with the fact that azithromycin inhibits mTOR, yet azithromycin is considered a useful immunomodulator drug for treating ME/CFS.
More on ketamine as a mTOR activator: this study says ketamine induces mTOR activation:
So with ketamine, as with leucine/glutamate, there seems to be an optimum dose, and if you go higher, you lose the effect.There is an inverted U-shape associated with ketamine-induced mTOR activation, with higher doses having no effect.
They also mention that:
other antidepressants, including 5-HT2C receptor antagonists, citalopram ... all increase mTORC1 levels
However, the SSRI, sertraline, and the TCA, imipramine, actually have anti-proliferative effects that are mediated by inhibition of mTOR
However, this idea that boosting mTOR may be helpful in ME/CFS does not fit in with the fact that azithromycin inhibits mTOR, yet azithromycin is considered a useful immunomodulator drug for treating ME/CFS.