

Ema
Senior Member
- Messages
- 4,729
- Location
- Midwest USA
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC64748/
More decreased dopamine and increased prolactin...clearly an influence on metabolism.
More decreased dopamine and increased prolactin...clearly an influence on metabolism.
That plasma leptin concentration declined gradually in anesthetized male rats during surgery led us to hypothesize that leptin secretion was under neural control (22). To assess the neural control of leptin, we used another type of stress, namely, the inflammatory stress induced by LPS that is known to increase leptin release (16). Furthermore, it was shown that LPS increased catecholamine levels in the CNS (9, 11) and in the periphery (10) and also activated the hypothalamic–pituitary–adrenal axis (5, 6). Therefore, we considered the present paradigm as a suitable model to assess the neural control of leptin.
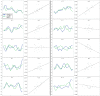
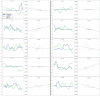
Our previous results had shown that LPS evoked a rapid and long-lasting increase in plasma leptin concentrations, with the first increase obtained within 10 min and a plateau existing from 2 to 6 h. The current results show that at 6 h there was a highly significant increase in leptin mRNA in epididymal fat pads induced by LPS. The early release of leptin within 10 min could not have occurred by new synthesis of leptin and, instead, must be caused by release of stored leptin that has been found in pinocytotic vesicles in adipocytes (26). Therefore, presumably, LPS acts on its receptors within the brain or receptors on afferent neurons, such as vagal afferents, to activate neural or hormonal mechanisms that evoke exocytosis of leptin-containing vesicles, which accounts for the initial elevation of plasma leptin. This is followed by induction of leptin mRNA, which stimulates leptin synthesis, contributing to the release that occurs later along with that of preformed leptin.
As in our previous results with placement of jugular catheters in anesthetized rats (22), plasma leptin decreased in the current experiments. Moreover, anesthesia also decreased LPS-induced plasma leptin release in the same period, providing further support for our concept that leptin is under neural control. Surprisingly, ketamine anesthesia either alone or in the presence of LPS provoked a rebound in plasma leptin levels after 120 min that reached concentrations similar to or even greater than those present in the LPS-treated rats. We hypothesize that this rebound may be caused by decreased negative feedback of the depressed plasma leptin concentrations during anesthesia, which, at the termination of anesthesia, act centrally to stimulate leptin release.
To understand further the possible role of the sympathetic nervous system in LPS-induced leptin release, we studied the effects of α- and β-adrenergic agonists and antagonists on the response to LPS. Injection of the β-adrenergic agonist, isoproterenol, slightly but significantly decreased plasma leptin concentrations and, in the presence of LPS, largely blunted the LPS-induced increase in plasma leptin concentrations. Recently, it has been shown that there is noradrenergic innervation not only of brown but also of white fat (27). That propranolol increased baseline concentrations of plasma leptin is consistent with the hypothesis that there is β-adrenergic inhibitory tone depressing leptin release during resting conditions. However, in the presence of LPS, propranolol slightly but not significantly decreased LPS-induced leptin release, suggesting that the inhibitory β tone under resting conditions was not present after injection of LPS. Therefore, our data suggest that either circulating epinephrine and/or norepinephrine or norepinephrine released from noradrenergic terminals may inhibit leptin release by acting on β-adrenergic receptors present on cell membranes of the adipocytes (28).
Phentolamine, the α-adrenergic antagonist, induced a rapid and highly significant increase in plasma leptin either alone or in the presence of LPS, suggesting that there is a strong inhibitory tone acting through the α-adrenergic receptors to inhibit not only basal but LPS-stimulated leptin release.
Recently, we found that prolactin stimulated leptin release and that α-bromoergocryptine, a dopaminergic-2 receptor agonist that inhibits prolactin release from the anterior pituitary gland, decreased plasma leptin concentrations (21), results that were obtained independently by Gualillo et al.(29). Moreover, we have shown previously that LPS increased prolactin release (5) in a similar experimental paradigm as that used here. Thus, it is likely that LPS-induced prolactin release increases leptin release by activation of prolactin receptors on adipocytes (30). Therefore, we studied the effect of α-bromoergocryptine, an inhibitor of prolactin release, alone or in the presence of LPS. As in the case of anesthesia, α-bromoergocryptine alone or in the presence of LPS decreased plasma leptin concentrations initially, but this decrease was followed by a later rebound of a lesser extent than that observed in the ketamine experiments. The rebound in both cases may be related to the initial lowering of plasma leptin, reducing its negative feedback. Then, as the drug or anesthesia dissipates, an increase in leptin release follows, leading to the rebound in plasma levels, which presumably is caused by increased prolactin release.
The decline in leptin in the bromocryptine-injected animals was much less than in the anesthetized animals, and the rebound was also less, perhaps because the lesser decrease in plasma leptin compared with that of anesthetized rats resulted in a lesser negative feedback of leptin in these animals.
We hypothesize that LPS acts on the CNS to inhibit the secretion of dopamine, removing the inhibition exerted by tuberoinfundibullar dopaminergic neurons on the secretion of prolactin (PRL) (see summary diagram, Fig. Fig.10).10). Therefore, the secretion of PRL is increased from the lactotropes. Thereafter, PRL circulates to the adipose tissue and, acting on its receptors (PRLr) on the adipocytes, increases the release of leptin that is stored in pinocytotic vesicles in the cytoplasma adjacent to the cell membrane. The sympathetic nervous system exerts a tonic inhibitory effect on leptin release—mediated predominantly by α-adrenergic and, to a lesser extent, by β-adrenergic receptors—that is still present and may be augmented by LPS.

")