

Annikki
Senior Member
- Messages
- 146
I study various autoimmune diseases. Today I'm focusing on endometriosis. I always thought that the way in which endometrial cells run amok in endometriosis was as if a retrovirus hijacked these cells.
Retroviruses have been implicated in cancer, because also in cancer you have normal cells behaving very abnormally and damaging towards the host. Viruses really don't care about their human hosts and, again, it makes little sense our own bodies would turn on us for the heck of it. I'm always amazed how ME has manifested in local epidemics. It also affects more women than men. For what it's worth, something is wrong in the bodies of ME patients and it goes against nature for body to merely sabotage itself. I imagine ERVs are relevant to ME, but unfortunately, ME research is still paltry because it has become a politicized disease.
Anyway I found an intriguing study about endometriosis which implicated endogenous retroviruses in the disease:
This study melds nicely with research which has found the placentas of mammals evolved using the genetic material of endogenous retroviruses. Endogenous retroviruses may have other actions in the uterus, which lead to endometriosis.
Maybe the body tolerates these ERVs because of their role in forming the placenta. The placenta forms from the endometrium. What could be going on with endo is the mechanisms which could and should dull ERV activity go faulty.
Maybe ERVs in endometrial cells get out of control and infect endometrial cells to use them to self-replicate their viral genome. If a endogenous retrovirus is dictating how endometrial cells replicate, you have an explanation as to how and why these cells start popping up in weird areas of the human body.
Retroviruses have been implicated in cancer, because also in cancer you have normal cells behaving very abnormally and damaging towards the host. Viruses really don't care about their human hosts and, again, it makes little sense our own bodies would turn on us for the heck of it. I'm always amazed how ME has manifested in local epidemics. It also affects more women than men. For what it's worth, something is wrong in the bodies of ME patients and it goes against nature for body to merely sabotage itself. I imagine ERVs are relevant to ME, but unfortunately, ME research is still paltry because it has become a politicized disease.
Anyway I found an intriguing study about endometriosis which implicated endogenous retroviruses in the disease:
Expression of Human Endogenous Gammaretroviral Sequences in Endometriosis
Abstract
Endogenous retroviruses (ERVs) probably originate from ancient germ cell infections by exogenous retroviruses. A high expression of retroviruses in reproductive tissue increases the risk of viral transmission to germ line cells. We therefore investigated the expression of human ERVs (HERVs) in normal endometrium, endometriosis, normal ovaries, and ovarian cancer. Four real-time PCRs (QPCRs) for HERV-E, HERV-I/T, HERV-H, and HERV-W, respectively, and an expression control gene were used. HERV-E RNA expression was significantly higher in endometriotic tissue (average, SD) than in normal endometrium (average, SD), both measured as ratios versus control gene expression and as. HERV-E and HERV-W RNA were higher in normal ovarian tissue than in ovarian cancer. This illustrates that HERV expression is not automatically higher in malignant tissues. The other HERV PCRs did not show expression patterns as distinctive as HERVE and HERV-W in the two kinds of reproductive tissue. A small number of candidate HERV-E loci from which the transcription took place were identified by sequencing of amplimers. The role of HERV-E and HERV-W in endometriosis merits further investigation.
https://www.liebertpub.com/doi/10.1089/aid.2006.22.551
This study melds nicely with research which has found the placentas of mammals evolved using the genetic material of endogenous retroviruses. Endogenous retroviruses may have other actions in the uterus, which lead to endometriosis.
Maybe the body tolerates these ERVs because of their role in forming the placenta. The placenta forms from the endometrium. What could be going on with endo is the mechanisms which could and should dull ERV activity go faulty.
Maybe ERVs in endometrial cells get out of control and infect endometrial cells to use them to self-replicate their viral genome. If a endogenous retrovirus is dictating how endometrial cells replicate, you have an explanation as to how and why these cells start popping up in weird areas of the human body.
Retroviruses facilitate the rapid evolution of the mammalian placenta
Edward B. Chuong
Abstract
The mammalian placenta exhibits elevated expression of endogenous retroviruses (ERVs), but the evolutionary significance of this feature remains unclear. I propose that ERV-mediated regulatory evolution was, and continues to be, an important mechanism underlying the evolution of placenta development. Many recent studies have focused on the co-option of ERV-derived genes for specific functional adaptations in the placenta. However, the co-option of ERV-derived regulatory elements has the potential to co-opt entire gene regulatory networks, which, I argue, would facilitate relatively rapid developmental evolution of the placenta. I suggest a model in which an ancient retroviral infection led to the establishment of the ancestral placental developmental gene network through the co-option of ERV-derived regulatory elements. Consequently, placenta development would require elevated tolerance to ERV activity, which in turn would expose a continuous stream of novel ERV mutations that may have catalyzed the developmental diversification of the mammalian placenta.
Keywords: conflict, co-option, endogenous retroviruses, evolution, evolvability, placenta, regulatory evolution
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4332834/
Retroviruses and the Placenta
DavidHaig
Retroviruses are often expressed in the placenta. Placental expression probably evolved to facilitate retroviral transmission from mother to offspring and from offspring to mother. In the process, the placenta became a site where retroviral genes were ‘domesticated’ to serve adaptive functions in the host, including the manipulation of maternal physiology for the benefit of the fetus. The evolutionary interplay between retroviruses and host defenses may have contributed to the remarkable diversity of form among mammalian placentas and to mechanisms of genomic imprinting.
Introduction
Infectious retroviruses possess an RNA genome that is reverse transcribed into double-stranded DNA, which is then inserted into the genome of a host cell as a provirus. Once inserted, the retrovirus replicates by transcription of new RNA genomes from the provirus or by DNA replication of the provirus as part of the host genome. Because of a long history of retroviral insertions into germ cells, mammalian genomes contain a substantial proportion of retroviral sequences in various stages of mutational decay [1].
Retroviruses appear to have a particular ‘affinity’ for placentas. Retroviral particles and mRNAs are often observed in placentas 2, 3, 4 and several genes use retroviral promoters to produce placenta-specific transcripts [5]. Domesticated retroviral envelope proteins (‘syncytins') promote the fusion of mononucleate trophoblast cells to form a syncytial layer at the maternal–fetal interface of primates and rodents 6, 7, 8, 9, and are suspected of performing a similar function in ruminants, lagomorphs, and carnivores 10, 11, 12. Most remarkably, the syncytins of each of these taxa have been recruited from different retroviral families.
Before the discovery of syncytins, virologists had proposed an important role for retroviruses in placental evolution, with the facilitation of trophoblast fusion and the suppression of maternal immune responses suggested as new functions provided by retroviral genes 3, 13, 14, 15. This review complements these earlier hypotheses by proposing a reason why retroviruses are often expressed in the placenta. Specifically, the origin of a placenta created new opportunities for retroviral transmission from mother to offspring and from offspring to mother (Figure 1). These new routes of contagion were exploited by retroviruses able to replicate in the trophoblast. Hosts were selected to block each new vulnerability and retroviruses to evade each new host defense in an intermittent ‘arms race.’ As a corollary, the placenta became a preferential site for the cooption of retroviral genes for adaptive host functions, including the suppression of infectious retroviruses. Before making these arguments, I will review briefly the selective forces acting on retroviral lineages.
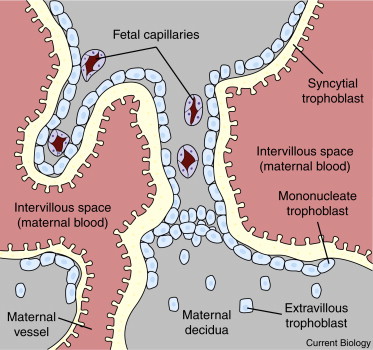
Figure 1. The maternal–fetal interface of the human placenta.
Extravillous trophoblast invades the maternal decidua and opens maternal blood vessels. Maternal blood flows through the intervillous space of the placenta. The intervillous space is lined by a layer of syncytial trophoblast that is maintained by fusion of underlying mononucleate trophoblasts with the syncytium, a process mediated by retroviral-derived syncytins. Retroviruses in maternal blood can potentially infect syncytial trophoblast and retroviruses produced in syncytial trophoblast can be released into the maternal circulation.
Retroviral Evolutionary Dynamics
A distinction is commonly made between endogenous and exogenous retroviruses. An endogenous retrovirus (ERV) is incorporated into the host germline, and has copies in every cell of an infected host, whereas an exogenous retrovirus is present in the genome of somatic cells only. Endogenous viruses can be transmitted from parent to offspring via gametes without ever having to infect a new cell, but an exogenous virus must repeatedly jump from older to younger somata to be maintained within a population. Some viral lineages, however, contain endogenous and exogenous elements that are indistinguishable, apart from their history of recent insertions. An exogenous virus becomes an endogenous virus, without changing its sequence or properties, when it infects a germ cell and an endogenous virus becomes an exogenous virus when one of its somatic copies produces an infectious particle that infects another somatic cell of the same or a different host.
The sense in which ERVs are ‘selfish genetic elements’ is subtle. Successful insertions select for elements that are able to copy themselves and move to new sites. Thus, adaptations that enhance transposition accumulate and are maintained in lineages that repeatedly change their chromosomal location. However, once an element resides at a new locus it is subject to the same selective forces as any other piece of chromosomal DNA [16]. Selection at the new site does not favor the ability to transpose, rather the reverse. Haplotypes on which active elements reside will be eliminated rapidly from a population if viral replication is associated with significant costs to organism fitness. Insertions that survive this selective filter are subject to selection of variants that enhance host fitness and to mutations that degrade former viral functions no longer subject to selection. Thus, most elements are subject to inexorable degradation of their ability to transpose [17]. In the simplest case, homologous recombination between the flanking long-terminal repeats (LTRs) of a retroelement generates a ‘solo LTR’ with deletion of all intervening material [18]. Germlines retain active elements only to the extent that some lineages transpose to new loci faster than elements are eliminated or domesticated.
Undoubtedly, many more retroviruses have moved into the germline than have left traces in sequenced genomes because of host-level selection against new insertions. Each insertion occurs on a single chromosome and is therefore initially a rare variant at its locus. If the element causes substantial costs to organism fitness, then it is doomed at that locus, although it may leave transposed descendants at other sites before its own extinction. For an insertion to become common, its effects must be nearly neutral and its haplotype increase in frequency by drift or hitchhiking (‘luck'), or the insertion must confer an advantage on its haplotype and be subject to a selective sweep. Therefore, actively-transposing elements will usually be rare at each particular locus because they are costly to hosts, whereas fixed elements are likely to be nearly neutral or beneficial to hosts [19].
Host defenses have evolved to control costs associated with active transposition 20, 21. Effective defenses inactivate most newly inserted elements. This increases the proportion of insertions that are nearly neutral and thereby promotes the accumulation of retroelements, or their remnants, in the genome [22]. Some of these elements may increase in frequency because they confer a benefit on host fitness by interfering with the transposition of active elements, including their own progenitors 23, 24.
Placental Contagion
Retroviruses gain entry to new host cells when viral coat proteins encoded by envelope (env) genes bind to cell-surface receptors. The coding sequence of an env gene is maintained under one of two conditions: either it belongs to a retroviral lineage that has continued to move between cells or the env protein has acquired a function beneficial to host fitness. Several ERV families expressed in the human placenta contain members with intact env genes [25], suggesting that the principal mechanism of proliferation within these families has involved reinfection, i.e. movement between cells by elements that encode their own env [19]. Placental expression of ERVs would be explained if such activity facilitated entry of the ERVs' progenitors into the germline. For example, placental expression of an ERV might induce immunological tolerance to its subsequent mobilization in other tissues [9]. A simpler hypothesis is that ERVs are expressed in the trophoblast because their immediate progenitors transposed into the germline from the trophoblast (or from the trophoblast via other somatic cells).
The close apposition of uterine and placental tissues creates a site for viral transmission from mother to fetus. By this path, a heterozygous ERV in the mother could potentially colonize all of a mother's offspring, not just the 50% that inherit the ERV by Mendelian means. For this to be an effective route of ongoing contagion, viruses transmitted from mother to placenta must sometimes re-infect somatic or germ cells of the fetus (or mother) before the placenta is discarded at delivery. Retroviruses are known to use this route: HIV-1 can be transmitted from mother to fetus across the placenta [26] and endogenous Jaagsiekte Sheep Retroviruses (enJSRVs) expressed in the uterine epithelium of ewes infect transplanted cattle embryos (Box 1) [27]. Transplacental infection is analogous to vertical transmission of retroviruses in breast milk 23, 28.
Box 1
Endogenous retroviruses of sheep.
Interactions among Jaagsiekte sheep retrovirus (JSRV), enzootic nasal tumor virus (ENTV), and related endogenous retroviruses (enJSRVs) illustrate the complex ecology of infectious and domesticated retroviruses. JSRV and ENTV cause tumors of the respiratory epithelia of sheep and are spread by respiratory aerosols 42, 43 whereas enJSRVs are expressed on both sides of the maternal–fetal interface by trophoblast and uterine epithelium [44]. Several enJSRVs have incorporated into the sheep genome over the past 5–7 million years, presumably from a pool of low-frequency infectious viruses of which they are the sporadic record. The most recent insertions, many of them polymorphic, form a clade with JSRV and ENTV [45].
During ovine pregnancy, binucleate trophoblast cells (BNCs) fuse with uterine epithelial cells to produce a syncytial plaque populated by placental and maternal nuclei (Figure 2) [46]. Multiple enJSRV env genes are expressed in BNCs, syncytial plaques, and the uterine epithelium [44]. Suppression of env proteins on both sides of the maternal–fetal interface causes trophoblast abnormalities and abortion of most pregnancies [10].
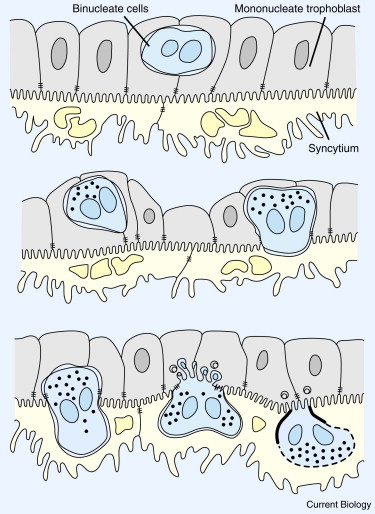
Figure 2. Formation of the syncytial plaque of ovine placentas.
Binucleate cells form in the layer of mononucleate trophoblast and fuse with uterine epithelial cells to form a syncytium containing nuclei from both generations. Syncytins derived from retroviral env genes are suspected of mediating these cell fusions. Modified after [46].
enJSRV-encoded env proteins interfere with JSRV entrance into cultured cells by receptor interference [47] and a gag protein encoded by enJS56A1, a duplicated ERV, interferes with JSRV exit from cultured cells 45, 48. enJSRVs are highly expressed in trophoblast and uterine epithelium but not at appreciable levels in the pulmonary and nasal epithelia where JSRV and ENTV are expressed [49]. Therefore, JSRV and ENTV are unlikely to be the targets of enJSRV-encoded interference. In fact, enJSRVs may tolerize adult sheep to JSRV and ENTV, explaining the absence of an antibody response to infections by these viruses [47]. Domesticated enJSRVs probably target infectious enJSRVs expressed in the placenta or uterus. One ERV, enJSRV-26, has been detected in a single sheep and is thus a strong candidate for an infectious ERV recently integrated into the germline. Significantly, a mutation in the signal peptide of its env gene allows enJSRV-26 to evade restriction by the enJS56A1 gag protein [50].
Retroviruses could also be transmitted across the placenta in the reverse direction, from offspring to mother. In this scenario, placental expression allows ERVs to colonize new sites in maternal genomes. An ERV expressed in the trophoblast could release particles or microvesicles that infect maternal tissues (or the tissues of litter-mates via the maternal circulation). Insertions in somatic cells of the mother could be a way-station for infection of her oocytes (with endogenous transmission to future offspring) and for various routes of exogenous transmission (including transmission across future placentas to future offspring).
Male and female germlines are different environments from the perspective of ERVs and related retroelements. Spermatogonial stem cells, for example, continue to divide throughout adult life, such that a new insertion of an exogenously acquired virus can be transmitted to an indefinite number of sperm. By contrast, female germ cells enter meiosis during embryonic development. These differences may create distinct vulnerabilities to retroviral proliferation in male and female germlines and, as a consequence, different repertoires of host defenses. From the host perspective, the benefits of suppressing retroviral transmission from trophoblast to mother is much weaker for fathers than for mothers, especially when mothers produce future offspring with new partners, because any costs of retroviral colonization of mothers' germlines or somata will have little effect on fathers' future fitness. Therefore, one might expect relaxed selection on paternal germlines to suppress placental retroviruses transmitted via sperm but stringent selection on maternal germlines to suppress the same retroviruses transmitted via eggs. These considerations raise the question whether some ERVs are expressed at higher levels in trophoblast when paternally inherited than when maternally inherited (i.e., whether ERVs exhibit imprinted expression).
Genome Defense and Genomic Imprinting
Genomic imprinting refers to the process by which some genes are expressed differently depending on whether they are inherited from mother or father. Host defenses against selfish genetic elements have been proposed to explain the evolution of genomic imprinting 29, 30, 31, 32. Such proposals are motivated by three principal lines of evidence. First, there is considerable overlap in the molecular machinery responsible for suppression of transposable elements (TEs) and for imprinted gene expression 29, 33. Second, some families of repetitive sequences show different epigenetic modifications on maternal and paternal transmission 34, 35. Third, some imprinted genes are derived from retroviruses or are retroposed host genes that have moved to new sites using retroelement-encoded mechanisms 5, 36.
Barlow [29] noted that DNA methylation both inactivates ‘foreign’ DNA and controls imprinted gene expression. She proposed that the ancestral function of DNA methylation was host defense and that imprinted genes contain sequences that are subject to methylation because they look like foreign DNA. McDonald and colleagues [30] similarly proposed that imprinted genes possess features that cause them “to be perceived as ‘foreign’ nucleic acids by the host cellular defense systems that target TE and viral transcripts”. These hypotheses appear to argue that defense is the primary function of DNA methylation and that imprinting is an incidental side-effect for genes that happen to look foreign.
Phylogenetic comparisons have been interpreted as strongly supporting the genome-defense hypothesis for the evolution of imprinting [31] and as providing “direct evidence that retrotransposon insertion can drive the evolution of genomic imprinting in mammals” [37]. These conclusions are based on evidence that marsupial and eutherian genomes contain more LTR elements than monotreme genomes [31] and that two paternally-expressed imprinted genes, PEG10 and RTL1, have been derived from sushi-class retrotransposons [36]. PEG10 is absent from monotreme genomes but present in marsupial and eutherian genomes [37] whereas RTL1 is restricted to eutherian genomes [38]. PEG10 is seen as having been imprinted at the moment of insertion in the genome because it was recognized as foreign by host defense mechanisms 31, 37. But this does not explain why silencing should be maternal-specific at PEG10 (but paternal-specific at other loci); why other insertions of similar elements are unimprinted [36]; how the initial insertion on a single chromosome spread to fixation; and, why imprinting has been maintained at this locus since the common ancestor of marsupial and eutherian mammals. All these questions are the province of adaptive hypotheses such as the parental conflict hypothesis [39].
Genome-defense hypotheses have hitherto had little to say on the defining feature of genomic imprinting, namely why defense mechanisms should be sex-specific. In the previous section, I suggested that ERVs might exhibit preferential paternal expression in trophoblast because of relaxed selection in paternal germlines to reduce the spread of ERVs from placentas to mothers. Similar arguments can be made for epigenetic modifications of other kinds of selfish genetic elements. Whether these modifications are predicted to favor maternal or paternal expression will depend on the details of how particular elements increase their copy number in male and female germlines, and whether the modifications are adaptations of the host or parasite. Sex-specific modifications of gametes may sometimes reflect sex-specific adaptations of hosts, because the two germlines have different vulnerabilities to selfish genetic elements, and sometimes reflect sex-specific adaptations of the elements to exploit these sex-specific vulnerabilities. As an added complication, conflict between maternal and paternal genomes over imprinted gene expression may result in defensive flaws that are exploited by selfish elements.
The genome-defense and parental conflict hypotheses have sometimes been presented as rival explanations for the evolution of genomic imprinting. Academic disputes are futile if the supposedly competing camps address different questions. In particular, questions of evolutionary history and mechanism are complementary to questions of adaptive function [40]. The defense hypothesis addresses the ancestral function of imprinting mechanisms whereas the conflict hypothesis is concerned with questions of why an initially rare imprinted allele should spread to fixation (selective origin) and why imprinting persists for long evolutionary periods at imprinted loci (selective maintenance). Hypotheses about the adaptive function of imprinting presuppose a source of ‘imprinted’ variation, otherwise there is nothing on which natural selection can act. Different defense mechanisms in male and female germlines provide a plausible source of such variation.
Conclusion
Mother and offspring come into intimate contact at the placenta, which is a potential route for transmission of pathogens between the generations. The placenta is also a site across which resources are transferred to the developing fetus and from which hormones and other factors are released into the mother's circulation to influence maternal metabolism for fetal benefit. As such, a fetus resembles a parasite engrafted on a maternal host [41]. Natural selection may have coopted adaptations of retroviruses, for their parasitic existence, to help fetuses ‘parasitize’ their mothers.
https://www.sciencedirect.com/science/article/pii/S0960982212006410
Last edited: