

View the Post on the Blog
Simon McGrath reports on the new study that indicates low-grade encephalitis in ME/CFS ...
A small study with just nine patients has captured the attention of patients and researchers alike after reporting direct evidence of inflammation in the brain of ME/CFS patients. The finding was one of the highlights picked out by Professor Anthony Komaroff in his IACFS/ME conference round up.
Back to the future
What makes this study so fascinating is that it provides tantalising evidence supporting not only of current views that inflammation in the brain is central to understanding the disease, but also of Melvin Ramsay's original name of 'myalgic encephalomyelitis'.
Encephalomyelitis is inflammation of the brain and spinal column, and critics of the name pointed to the lack of direct evidence for inflammation of either. This study only looked at the brain, not the spinal column (so could only find encephalitis), but the immune cells found to be activated in the brain are also present in the spinal column.
The study

Dr. Yasuyoshi Watanabe
To see if there is immune activation in the brain, researchers need to look inside the brain -- which is not so easy if you want patients to still be alive when your study is done.
The scientists in this study, led by Dr. Yasuyoshi Watanabe from the RIKEN institute in Japan, used PET & MRI imaging to peer into the brain.
What make this study work is the use of tiny quantities of a radioactive tracer that binds to specific proteins that appear on activated microglia (the main immune cells of the brain) but crucially doesn't bind to non-activated microglia. The marker also binds to activated astrocytes, which play an immune role in the brain. The brains of nine ME/CFS patients meeting both Fukuda and International Consensus Criteria were compared with those of 10 healthy controls.
The results showed that neuroinflammation markers were higher for patients than controls across many brain areas including the thalamus, the pons and the midbrain. They also found that the severity of symptoms correlated with the degree of inflammation in multiple brain regions, particularly for cognitive functioning.
It was the correlation between a biological finding -- neuroinflammation -- and clinical problems that Komaroff found so exciting about this work, because it suggests a biologically plausible explanation for the symptoms of ME/CFS:
"[If replicated] it would, for me, say that there is a low-grade, chronic encephalitis in these patients, that the image we clinicians have of encephalitis as an acute and often dramatic clinical presentation that can even be fatal has -- may have -- blinded us to the possibility that there may be that long-lasting -- many years long -- cyclic chronic neuroinflammation is underlying the symptoms of this illness."
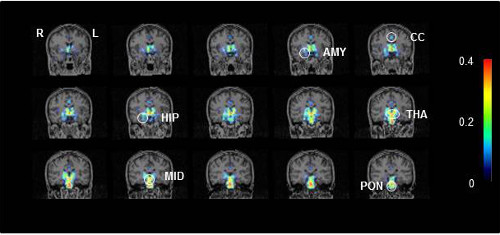
Representative PET scans showing activated microglia in a CFS/ME patient.
Key to brain regions: AMY, amygdala; CC, cingulate cortex; HIP, hippocampus; MID, midbrain; THA, thalamus; and PON: pons.
Photo credit: Image courtesy of RIKEN
Intriguingly, the midbrain, thalamus and amygdala -- all regions where cognitive problems correlate with neuroinflammation -- are also all part of neural circuits involved in awareness, arousal and attention. Concentration problems are typical of ME/CFS, and one of the problems found most consistently in laboratory testing.
Harvard Professor Tony Komaroff on these PET findings, and their potential importance
Starts at 30' 10", Q&A re encephalomyelitis @ 37'.
Replication needed
While tantalising, these findings are far from conclusive, as the authors acknowledge. The study has only nine patients, albeit diagnosed with ICC criteria. The tracer used to identify activated immune cells produces a very 'noisy' signal, giving rather indistinct readings, and the overall level of neuroinflammation was relatively low.
Although cognitive issues correlated with neuroinflammation in several areas, generally other symptoms, including fatigue, did not significantly correlate with inflammation.
There was almost no sign of inflammation in the prefrontal cortex, the region of the brain most involved in higher cognitive functions, that might be expected to be a problem in ME/CFS. And there was a potential technical weakness in the way the study was run.
Commenting on the neuroinflammation, Komaroff emphasised the need for replication:
Hopefully independent groups will try to replicate this finding too - and in the U.K., Dr. Charles Shepherd of the ME Association has already said it would welcome applications to fund a replication attempt.
Microglia -- key to ME/CFS?
So neuroinflammation -- specifically activation of microglia -- correlates with cognitive problems, but how might microglial activation cause the problem?
The most plausible answer is through what is termed 'sickness behaviour' -- a characteristic set of responses to infection, including fatigue, malaise joint and muscle pain and problems concentrating -- which might just sound familiar to ME/CFS sufferers. ('Sickness behaviour' is a lousy name for biological phenomenon, as Dr. Dan Peterson has noted).
Microglia are known to play a key role in regulating sickness behaviour, and that's a big reason this study has attracted so much attention in ME/CFS.
 'Sickness Behaviour' is driven by biology: infection leads to a rise in pro-inflammatory cytokines in the blood, triggering activation of brain microglia and their production of cytokines. This triggers sickness behaviour.
'Sickness Behaviour' is driven by biology: infection leads to a rise in pro-inflammatory cytokines in the blood, triggering activation of brain microglia and their production of cytokines. This triggers sickness behaviour.
The fatigue, malaise, problems concentrating, etc., of sickness behaviour help us survive an infection by forcing us to rest so our body can devote all its resources to the energy-greedy immune system.
However, sickness behaviour is normally a short-lived response to an acute infection, designed to temporarily divert resources to ensure a swift recovery. If that doesn't happen, e.g., if there is a chronic infection, or the process goes wrong, for instance, if microglia remain activated after an infection has been cleared, then sickness behaviour can itself be a problem. ME/CFS may be an example of this.
Cytokines in the spotlight
Cytokines are a key trigger for sickness behaviour, and researchers have often found elevated cytokines in patients, but the findings have been inconsistent and in small studies. The new studies reported on by Dr. Jose Montoya at the Stanford conference and Dr. Mady Hornig at the IACFS/ME conference are helping to firm up these findings in huge cohorts.
Probably the most important piece of work on the role of sickness behaviour -- and cytokines -- in ME/CFS came from the landmark "Dubbo" studies.
The researchers found that about 12% of those with glandular fever and two other infections developed CFS after six months. And crucially, what predicted the length of the illness (and chance of developing CFS) wasn't psychological factors, but the severity of the initial 'acute illness', or sickness behaviour.
The researchers also showed that those with more active genes for the pro-inflammatory cytokine Interferon-gamma had a more severe sickness behaviour (and longer illness) than those with regular versions, linking cytokine response to sickness behaviour and ME/CFS.
The Dubbo study did not look at inflammation in the brain, but the authors did speculate that the cause of CFS could be long-term activation of microglia and astrocytes. And that is exactly what was found in this new PET imaging study.
As with all research findings, replication is essential, and a new version of the Dubbo study is currently under way in Sydney, Australia.
The new imaging study from Japan has found provisional evidence of activated astrocytes and microglia cells (both types of glial cell) in the brain of ME/CFS patients. This is support for the suggestion from the Dubbo team that ME/CFS develops from certain infections as a result of activation of brain microglia.
Dr. Michael VanElzakker's recent vagus nerve infection hypothesis also features glial cells heavily. And recently Professor Hugh Perry, who has studied microglial cells in neurodegenerative diseases such as Parkinson's disease, proposed that primed microglia and sickness behaviour lie at the heart of ME/CFS.
Neuroinflammation and Sickness Behaviour the final common path in ME/CFS?
It may prove to be that 'neuroinflammation' -- i.e., activated microglia in the brain/spinal column -- is a common endpoint of numerous triggers, including glandular fever (EBV), other infections, vaccines -- or even, as Dr. Lipkin has proposed, disturbances in the microbiome.
Discovering if this is the case -- and firming up the finding of neuroinflammation is key -- could be a big step forward in understanding and then treating ME/CFS. And those it is still very early days, it is possible this approach could eventually show that Dr Ramsay was right about 'encephalomyeltitis'.
Watch out for a new blog on sickness behaviour, microglia, cytokines and their role in ME/CFS, coming soon.
Phoenix Rising is a registered 501 c.(3) non profit. We support ME/CFS and NEID patients through rigorous reporting, reliable information, effective advocacy and the provision of online services which empower patients and help them to cope with their isolation.
There are many ways you can help Phoenix Rising to continue its work. If you feel able to offer your time and talent, we could really use some more authors, proof-readers, fundraisers, technicians etc. We’d also love to expand our Board of Directors. So, if you think you can help in any way then please contact Mark through the Forums.
And don’t forget: you can always support our efforts at no cost to yourself as you shop online! To find out more, visit Phoenix Rising’s Donate page by clicking the button below.

View the Post on the Blog
Simon McGrath reports on the new study that indicates low-grade encephalitis in ME/CFS ...
A small study with just nine patients has captured the attention of patients and researchers alike after reporting direct evidence of inflammation in the brain of ME/CFS patients. The finding was one of the highlights picked out by Professor Anthony Komaroff in his IACFS/ME conference round up.
Back to the future
What makes this study so fascinating is that it provides tantalising evidence supporting not only of current views that inflammation in the brain is central to understanding the disease, but also of Melvin Ramsay's original name of 'myalgic encephalomyelitis'.
Encephalomyelitis is inflammation of the brain and spinal column, and critics of the name pointed to the lack of direct evidence for inflammation of either. This study only looked at the brain, not the spinal column (so could only find encephalitis), but the immune cells found to be activated in the brain are also present in the spinal column.
The study
Dr. Yasuyoshi Watanabe
The scientists in this study, led by Dr. Yasuyoshi Watanabe from the RIKEN institute in Japan, used PET & MRI imaging to peer into the brain.
What make this study work is the use of tiny quantities of a radioactive tracer that binds to specific proteins that appear on activated microglia (the main immune cells of the brain) but crucially doesn't bind to non-activated microglia. The marker also binds to activated astrocytes, which play an immune role in the brain. The brains of nine ME/CFS patients meeting both Fukuda and International Consensus Criteria were compared with those of 10 healthy controls.
The results showed that neuroinflammation markers were higher for patients than controls across many brain areas including the thalamus, the pons and the midbrain. They also found that the severity of symptoms correlated with the degree of inflammation in multiple brain regions, particularly for cognitive functioning.
It was the correlation between a biological finding -- neuroinflammation -- and clinical problems that Komaroff found so exciting about this work, because it suggests a biologically plausible explanation for the symptoms of ME/CFS:
"[If replicated] it would, for me, say that there is a low-grade, chronic encephalitis in these patients, that the image we clinicians have of encephalitis as an acute and often dramatic clinical presentation that can even be fatal has -- may have -- blinded us to the possibility that there may be that long-lasting -- many years long -- cyclic chronic neuroinflammation is underlying the symptoms of this illness."
Representative PET scans showing activated microglia in a CFS/ME patient.
Key to brain regions: AMY, amygdala; CC, cingulate cortex; HIP, hippocampus; MID, midbrain; THA, thalamus; and PON: pons.
Photo credit: Image courtesy of RIKEN
Harvard Professor Tony Komaroff on these PET findings, and their potential importance
Replication needed
While tantalising, these findings are far from conclusive, as the authors acknowledge. The study has only nine patients, albeit diagnosed with ICC criteria. The tracer used to identify activated immune cells produces a very 'noisy' signal, giving rather indistinct readings, and the overall level of neuroinflammation was relatively low.
Although cognitive issues correlated with neuroinflammation in several areas, generally other symptoms, including fatigue, did not significantly correlate with inflammation.
There was almost no sign of inflammation in the prefrontal cortex, the region of the brain most involved in higher cognitive functions, that might be expected to be a problem in ME/CFS. And there was a potential technical weakness in the way the study was run.
Commenting on the neuroinflammation, Komaroff emphasised the need for replication:
"If it were confirmed by multiple other investigators ... these data are consistent with [encephalitis], but I would feel more strongly if other labs using same technology came up with the same result."
The good news is that the authors of this study are already working on a new study using the same patients but with a newer and more sensitive tracer to pick up neuroinflammation. They will address the earlier technical issue, and to make the study more powerful they will also be looking at neurotransmitter activity in the brain, following up their previous findings of neurotransmitter abnormalities.
Hopefully independent groups will try to replicate this finding too - and in the U.K., Dr. Charles Shepherd of the ME Association has already said it would welcome applications to fund a replication attempt.
Microglia -- key to ME/CFS?
So neuroinflammation -- specifically activation of microglia -- correlates with cognitive problems, but how might microglial activation cause the problem?

The most plausible answer is through what is termed 'sickness behaviour' -- a characteristic set of responses to infection, including fatigue, malaise joint and muscle pain and problems concentrating -- which might just sound familiar to ME/CFS sufferers. ('Sickness behaviour' is a lousy name for biological phenomenon, as Dr. Dan Peterson has noted).
Microglia are known to play a key role in regulating sickness behaviour, and that's a big reason this study has attracted so much attention in ME/CFS.

The fatigue, malaise, problems concentrating, etc., of sickness behaviour help us survive an infection by forcing us to rest so our body can devote all its resources to the energy-greedy immune system.
However, sickness behaviour is normally a short-lived response to an acute infection, designed to temporarily divert resources to ensure a swift recovery. If that doesn't happen, e.g., if there is a chronic infection, or the process goes wrong, for instance, if microglia remain activated after an infection has been cleared, then sickness behaviour can itself be a problem. ME/CFS may be an example of this.
Cytokines in the spotlight
Cytokines are a key trigger for sickness behaviour, and researchers have often found elevated cytokines in patients, but the findings have been inconsistent and in small studies. The new studies reported on by Dr. Jose Montoya at the Stanford conference and Dr. Mady Hornig at the IACFS/ME conference are helping to firm up these findings in huge cohorts.
Probably the most important piece of work on the role of sickness behaviour -- and cytokines -- in ME/CFS came from the landmark "Dubbo" studies.
The researchers found that about 12% of those with glandular fever and two other infections developed CFS after six months. And crucially, what predicted the length of the illness (and chance of developing CFS) wasn't psychological factors, but the severity of the initial 'acute illness', or sickness behaviour.
The researchers also showed that those with more active genes for the pro-inflammatory cytokine Interferon-gamma had a more severe sickness behaviour (and longer illness) than those with regular versions, linking cytokine response to sickness behaviour and ME/CFS.
The Dubbo study did not look at inflammation in the brain, but the authors did speculate that the cause of CFS could be long-term activation of microglia and astrocytes. And that is exactly what was found in this new PET imaging study.
As with all research findings, replication is essential, and a new version of the Dubbo study is currently under way in Sydney, Australia.
The new imaging study from Japan has found provisional evidence of activated astrocytes and microglia cells (both types of glial cell) in the brain of ME/CFS patients. This is support for the suggestion from the Dubbo team that ME/CFS develops from certain infections as a result of activation of brain microglia.
Dr. Michael VanElzakker's recent vagus nerve infection hypothesis also features glial cells heavily. And recently Professor Hugh Perry, who has studied microglial cells in neurodegenerative diseases such as Parkinson's disease, proposed that primed microglia and sickness behaviour lie at the heart of ME/CFS.
Neuroinflammation and Sickness Behaviour the final common path in ME/CFS?
It may prove to be that 'neuroinflammation' -- i.e., activated microglia in the brain/spinal column -- is a common endpoint of numerous triggers, including glandular fever (EBV), other infections, vaccines -- or even, as Dr. Lipkin has proposed, disturbances in the microbiome.
Discovering if this is the case -- and firming up the finding of neuroinflammation is key -- could be a big step forward in understanding and then treating ME/CFS. And those it is still very early days, it is possible this approach could eventually show that Dr Ramsay was right about 'encephalomyeltitis'.
Watch out for a new blog on sickness behaviour, microglia, cytokines and their role in ME/CFS, coming soon.
Phoenix Rising is a registered 501 c.(3) non profit. We support ME/CFS and NEID patients through rigorous reporting, reliable information, effective advocacy and the provision of online services which empower patients and help them to cope with their isolation.
There are many ways you can help Phoenix Rising to continue its work. If you feel able to offer your time and talent, we could really use some more authors, proof-readers, fundraisers, technicians etc. We’d also love to expand our Board of Directors. So, if you think you can help in any way then please contact Mark through the Forums.
And don’t forget: you can always support our efforts at no cost to yourself as you shop online! To find out more, visit Phoenix Rising’s Donate page by clicking the button below.
View the Post on the Blog
Last edited by a moderator:
")