

necessary8
Senior Member
- Messages
- 134
EDIT 17/09/2018
Sadly, I have to inform you that I've found a critical flaw in this hypothesis. There are still many pieces to it which I would consider valuable ideas, but the whole no longer works together.
I have stated that "after activating the A1 receptors, a significant portion of this adenosine is actually brought into the cell by CNT2 transporters, and proceeds to activate AMPK. The exact mechanism by which it accomplishes that is not known, but my speculation is that it’s a different mechanism from the one AMP uses, and therefore serves as a fallback activation route in muscle exertion." - this is not fully correct. The adenosine is transported into the cells, and does activate AMPK, but the mechanism is not unknown. The adenosine turns into AMP, which activates AMPK in the usual manner. Meaning, this cannot be an alternative, fallback activation route for AMPK in contracting skeletal muscle. Unfortunately, this is one of the very few places where an error like this means the whole hypothesis falls apart. The two halves of this hypothesis - the systemic part with the anti-beta2 antibodies, and the intracellular part, with the phosphatidic acid - no longer fit together, as they were tied by this idea of adenosine constituting a fallback activation route.
This is fully my mistake, I missed an important part in one of my sources. I apologize terribly for wasting your time. The irony of working on this for half a year only to discover a critical flaw days after going public, would be funny to me if I wasn't so embarrassed by this.
I am leaving this post here, but the untrue statements as well as my ideas directly stemming from this misunderstanding, will be marked red.
The Adenosine - Phosphatidic Acid Hypothesis
Foreword
Hello everyone, my name is Matt, you may remember me as the guy from the CD39 hypothesis (if not: Part1, Part2). Today, I want to present you with something better, the result of my ponderings since that time. It attempts to tie together metabolism, autoimmunity, gut dysbiosis, AMPK, mTOR, ATP production, and explain all the major symptoms, as well as the metabolomics results. I call it the Adenosine - Phosphatidic Acid hypothesis, or the Adn-PA hypothesis for brevity.
Before you start reading, I have to warn you of two things. First, I decided that I will not include explanations for anything that you can easily google by yourself (eg what is AMPK and what it does), because honestly I had no energy left for that. And second, this hypothesis is complex. It is comprised of a dozen hypothetical feedback loops that all intertwine and interact with each other. If you wish to fully understand it, I would recommend reading while looking at the at the diagrams provided in Addendum 1 and Addendum 2, following the pathways in your mind, and perhaps also re-reading it a few times.
If you wish to see a summary first, before reading the whole thing, the diagram in Addendum 1 can serve as such.
The thing itself
Let’s start with the autoantibodies to beta-2 adrenergic receptors. They were discovered in about 30% of ME/CFS patients by Carmen Shcheibenbogen[source], and subsequently confirmed by other researchers. In this theory I am assuming that the rest 70% of patients have other yet undiscovered autoantibodies that achieve a similar effect, or that the T cells are doing a similar thing (both of which have been hinted at by researchers), or perhaps that the effects I describe below have also other systemic mediators. Some ideas for that I will include at the end.
The origin of those antibodies, I imagine, is genetic predisposition for autoimmunity, coupled with a triggering event which flipped some key epigenetic switch, allowing it to first take place. We know that, for example, the Epstain-Barr virus can do this.[source] The reason for why those antibodies persist, and even increase with response to exercise (my speculation), as well as how they cause ME/CFS symptoms, will be the subject of this hypothesis.
Beta-2-ARs are one of the most prominent G protein coupled receptors. They couple mainly with Gs types, meaning they stimulate adenylyl cyclase to produce cAMP, which then activates Protein Kinase A.[source] This action can sometimes get very difficult to track, because its effects don’t just differ between cell types, they also differ between specific localization in the cell. Cells with G protein coupled receptors have individual “pockets” inside them with a specific receptor, a localized pool of cAMP, phosphodiesterases surrounding it, preventing from influencing other pockets, and localized Protein Kinase A attached to a specific AKAP (A Kinase Anchoring Protein). There is about 40-something types of those, and they strongly modulate what the attached Protein Kinase A does.[source1][source2][source3][source4] So in one single cell, a G protein coupled receptor on one end of the cell, and a second receptor on the other end of the cell, could have very different functions, despite being the exact same type of a receptor, because their PKAs are attached to different AKAPs and their cAMP pools are separated by PDEs.
Fortunately, there are some aspects that are common. Of particular interest to me, is the mechanism of desensitization of those receptors. When cAMP levels rise, this activates MRP transporters which bring a significant portion of the cAMP outside of the cell. The cAMP is then converted to AMP by a yet-to-be-classified ecto-phosphodiesterase, and then the AMP is turned into adenosine by ecto-nucleotidase CD73. The adenosine then stimulates the A1 receptors of the cell, which are also GPCRs, but couple primarily with Gi types, so they inhibit cAMP production by adenylyl cyclases. This closes the loop of a negative feedback cycle, suppressing cAMP production completely after 15-60 minutes of constant receptor stimulation. This effect has been observed in many CGRPs, including beta adrenergic receptors.[source1][source2][source3]
This mechanism is interesting for a few reasons. Firstly, the autoantibodies in ME/CFS cause gain of function of the beta2 receptors - they make them activate more. That should mean more extracellular adenosine. And yet, looking at the metabolomics data from Robert Naviaux, andenosie is actually one of the most decreased compounds. It is extremely low.[source] There are, obviously, other sources of adenosine in the bloodstream, but my impression is that GPCR desensitization is one of the largest ones, if not the dominant one. This leads me to believe, that what happens in ME/CFS patients, is the cells after prolonged constant stimulation of their beta adrenergic receptors, take compensative measures to offset this. Perhaps by suppressing the genetic expression of those receptors, or G proteins, or by uncoupling them somehow. I don’t know how exactly. But the effect is a diminished and very constant level of activation, that is not very responsive to the proper highs and lows of the actual ligand concentration. In the context of vascular smooth muscles, this can explain POTS and orthostatic intolerance very well, as it would mean the blood vessels can’t adapt their width to keep the blood pressure constant, when faced with changing physical conditions (like standing upright).
But I am more interested in what this does in skeletal muscle. Now, in skeletal muscle the beta adrenergic receptors have some role in potentiation of the muscle contractile force, so this might mean some limited muscle weakness, which does seem to happen to patients. But I believe that a much more important change occurs because of the diminished extracellular adenosine production, as the cAMP levels never rise very high. In healthy people, the activation of skeletal muscle beta adrenergic receptors, and the subsequent adenosine production would presumably occur mainly in exercise, as that is when blood noradrenaline levels are high, and when noradrenaline is released from surrounding neurons.[source1][source2] There are even studies directly showing higher adenosine production after exercise.[source] So in my hypothesis here, the patients have lower resting adenosine levels, but much more importantly, those levels do not rise as they would in a healthy person during exercise. And I believe I have solid grounds to suspect that this rise in extracellular adenosine has multiple important roles during muscle exertion, and is not simply a side product of GPCR desensitization.
First such role is anti-inflammatory. It has been observed in multiple immune cell types, such as dendritic cells and macrophages, that extracellular adenosine attenuates their activation, and has even been hypothesized to mediate the mechanism of their deactivation in immune resolution.[source1][source2][source3][source4] This is important because the immune system participates to some degree in the muscle repair following exercise.[source1][source2][source3][source4][source5] And my guess is that adenosine serves as a moderator, keeping the immune cells in check and preventing them from reacting in an autoimmune manner.
Another such moderator is probably IL6, the interleukin that is also produced by muscle during exercise.[source] And in many cell types it has been observed that activation of the adenosine receptor A2B elicits IL6 production and secretion.[source][source2][source3][source4] It’s not a far-fetched idea that the same would happen in muscle cells. And so, if adenosine is lower, IL6 is lower.
(Another such factor might be BDNF[source], which is sometimes found to be low in ME/CFS as well, but I haven’t explored this enough to write about it yet)
Without those immune moderators, which are normally present during exercise, ME/CFS patients’ immune systems, activated by the muscle exertion, are more likely to go overboard, and autoimmune. And proceed to produce even more anti-beta2 antibodies and/or T cells, closing the disease feedback loop. The first one at least. There is more.
The feeling of fatigue is facilitated by synergistic activation of ASIC, TRPV1 and P2X3 receptors on certain nociceptive neurons. The main receptor is ASIC, with TRPV1 and P2X3 working to sensitize it, to activate more easily. The metabolites required for this activation are lactic acid and extracellular ATP (both of which are released from muscle cells during exercise).[source1][source2] And it just so happens that extracellular cAMP attenuates this process.[source] Meaning, if those neurons activate in healthy people in presence of lots of extracellular cAMP, and that gets you normal fatigue, then it makes sense than in ME/CFS patients, with much lower cAMP efflux, they activate more strongly and easily, resulting in intense fatigue. Certain levels of LA and ATP are present there even at rest, so without the inhibitory action of cAMP they could very well be activating even at rest.
But I think there is yet another mechanism at play here. There is a study that I keep going back to, done by Vincent Lombardi’s team, where they tested antibodies in serum of ME/CFS patients against 125000 peptides with random amino acid sequences, to find what the antibodies are reacting to, even if it’s a new unknown antibody. The study didn’t find a very specific match, but they did find a short sequence that, if I understand correctly, antibodies in the great majority of patients react to. And after they did a search to find human proteins containing it, or a similar sequence, among the 30 best matches for it was the ASIC1 receptor.[source] The main receptor responsible for facilitating the feeling of fatigue. Personally, I find it unlikely for this to be a coincidence, so what I hypothesize is happening here, is that antibodies with this sequence bind to the ASIC receptors and sensitize them in a similar manner that eg. the P2X3 receptor does. This acts synergistically with the lower levels of extracellular cAMP to activate the fatigue neurons more easily and strongly.
I’m writing about this not only to explain why the patients have intense fatigue, but also because this can very well constitute another feedback cycle reinforcing the disease. As Alan Light pointed out to me, the nociceptive neurons, when activating, secrete certain peptides like CGRP[source], and Substance P[source], which can modulate the behaviour of immune cells.[source] Hence it is possible that the activation of fatigue neurons itself also reinforces the autoimmunity.
All of the above by itself constitutes a hypothetical disease mechanism that explains a lot of the symptoms, but what I really wanted this to do, is to explain the metabolomics results, which I still consider the most important data we currently have on ME/CFS.
To do that, I must first talk about Julia Newton’s research regarding AMPK. Her team demonstrated that in skeletal muscle cells cultured from ME/CFS patients, and stimulated for contraction, there is less AMPK activity than in cells from healthy subjects.[source] I find this discovery particularly important precisely because it is in skeletal muscle cells (myotubes), and not in PBMCs (I explained why here). I’ve come up with dozens of ways for an autoimmune mechanism to cause AMPK impairment in skeletal muscle, but none of them can explain these results for one reasons - the cells in this study were not myotubes taken directly from patients, they were cultivated in a dish from patients’ muscle satellite cells, which are kind of resident blueprints for myotubes, used to expand and repair them when need be. What this means is that the myotubes in the dish share only the genetic and epigenetic factors, and not other influences, with the myotubes in the patients. So all of the processes that I described above, even if present in patients’ skeletal muscles, would not translate to the muscle cells in the dish here.
Therefore, what I propose, is that the impairments in AMPK activation in skeletal muscle which were identified in this study, are strictly genetic or epigenetic, and that patients carry them their whole life, even before developing ME/CFS. This impairment itself is not enough to cause the disease, but it lies very important groundwork for it to take place.
The reason why I think this, is because I believe there exist fallback mechanisms that make sure AMPK is activated during skeletal muscle contraction, which are independent of the direct activation by AMP accumulating in the cell. Particularly, I think I’ve found one such mechanism, which relies on the extracellular adenosine created in muscle contraction. As it turns out, after activating the A1 receptors, a significant portion of this adenosine is actually brought into the cell by CNT2 transporters, and proceeds to activate AMPK.[source] The exact mechanism by which it accomplishes that is not known, but my speculation is that it’s a different mechanism from the one AMP uses, and therefore serves as a fallback activation route in muscle exertion.
So, I propose that people with (epi)genetic impairment of AMPK activation in skeletal muscle stay healthy and don’t develop ME/CFS, because they still have this fallback mechanism working. They only get ME/CFS after developing autoimmunity to beta adrenergic receptors, which leads to impaired adenosine production in the extracellular space surrounding skeletal muscle, and leads to loss of this fallback mechanism. The result is AMPK that doesn’t sufficiently activate when the muscles become active, leading to uptake of glucose and ATP synthesis not becoming appropriately upregulated during exercise. The Krebs cycle, glycolysis and oxidative phosphorylation have other regulation mechanisms independent of AMPK, so this probably isn’t enough of an impairment for cells to die, but is enough to elicit some changes in them.
What changes exactly? Lots of things, probably; most of which I don’t know about. It could also perhaps activate the fatigue neurons further - I am skeptical of this idea because I haven’t seen any research demonstrating that this is possible, but I’ve seen a lot of researchers hint at this, and it certainly would make a lot of evolutionary sense for the fatigue neurons to be able to react directly to the energy state of the muscles, so I’m keeping an open mind. (If you know of any research demonstrating such a pathway existing, pretty please send it my way)
But here I wanted to focus on one particular consequence, which I haven’t seen addressed before, namely the effect on the Kennedy (CDP-choline) pathway. This pathways synthesizes phosphatidylcholine from choline, using ATP for the process. And it becomes largely downregulated if the cell doesn’t have plentiful energy. This is actually fully independent of AMPK - the choline kinase somehow senses the ATP/AMP ratio directly.[source1][source2]
My hypothesis here is that after a long time of the Kennedy pathway being inhibited by the low energy status, the skeletal muscle cells of ME/CFS patients start running low on phosphatidylcholine. This is consistent with the metabolomics results - the main thing that the two large metabolomics studies of Robert Naviaux[source] and Ian Lipkin[source] agree on, is that phosphatidylcholines are decreased.
The main role of phosphatidylcholine is to be a building material for cell membranes, but I wanted to focus on a different role here.
Phospholipase D is an enzyme that breaks down phosphatidylcholine to make phosphatidic acid. And I’ve found some really interesting papers demonstrating the very important role that this reaction plays in the regulation of mTOR and AMPK.
mTOR is a master regulator of protein synthesis in cells, and it is regulated by the TSC1/TSC2 complex, which, when active, prevents Rheb from associating with GTP, and only GTP-loaded Rheb can activate mTOR. All the pathways which influence mTOR go through here, either activating or inhibiting TSC1/TSC2 to control what mTOR is doing. And it turns out that the de facto mechanism by which GTP-loaded Rheb activates mTOR involves Phospholipase D1. This phospholipase is activated by GTP-Rheb, producing phosphatidic acid, which in turn displaces the inhibiting protein FKBP38 from mTOR, allowing Rheb to bind to mTOR in its place and activate it. If Phospholipase D1 action is inhibited, this process cannot occur and mTOR cannot activate.[source1][source2]
Phospholipase D1 also has an equally important role with AMPK. One would think that since mTOR and AMPK have an antagonistic relationship, that Phospholipase D1 activity would inhibit AMPK, or the other way around, and there is indeed one paper claiming that.[source] But there is more and much more thorough evidence, that AMPK actually activates Phospholipase D1. Moreover, the resulting phosphatidic acid, by facilitating translocation of Raf1 to cell membrane, is the mechanism by which AMPK activates the ERK/MAPK cascade and upregulates glucose uptake in times of higher energy demand. (and probably, it’s also the mechanism by which Akt/PKB activates ERK/MAPK, since Akt lies upstream of TSC1/TSC2) If Phospholipase D1 is inhibited, AMPK cannot fully upregulate glucose uptake.[source1][source2][source3]
With all this said, I do not think Phospholipase D1 action is defective in ME/CFS. Rather, because of the aforementioned phosphatidylcholine deprivation, Phospholipase D1 runs out of substrate for breaking down into phosphatidic acid.
If this becomes the case, even if the prior impairment in AMPK activation was fairly minor, the lack of phosphatidic acid production further prevents AMPK action, not by directly inhibiting it, but by making it unable to upregulate glucose uptake properly in times of greater energetic demand, starving the Krebs cycle of fuel, which further inhibits the Kennedy pathway, and we have another feedback loop.
It also would strongly inhibit mTOR activation. But, interestingly enough, it’s possible that Akt would actually be upregulated here. This is because a particular kind of phosphatidylcholine - PC 20:4, is an Akt inhibitor[source], and with its production downregulated, Akt might activate more. This would lead to inhibition of FoxO factors, acting synergistically with the inhibited AMPK (AMPK also activates FoxO). This inhibition of FoxO could lead to downregulation of Sestrin 3, which is another AMPK activator, potentially furthering AMPK inhibition.[source]
But more importantly, FoxO3 is an important mediator of autophagy. While it is not directly stated anywhere in the literature, it is my understanding after reading it, that AMPK and mTOR act synergistically to repair damage done to the muscles during exercise. AMPK acts first, activating FoxO3, enabling autophagy, which removes all the damaged parts.[source1][source2] Then mTOR mediates synthesis of new proteins to replace them.[source1][source2] And elevated IL6 levels signal the satellite cells to help with this process also, providing new nuclei etc for the myotubes undergoing repair.[source]
So what I propose occurs in the muscles of ME/CFS patients, is metabolic stagnation, caused by AMPK and mTOR not activating properly in exercise, and also by IL6 not being secreted as much. Metabolic stagnation, meaning, old stuff isn’t broken down as fast, and new stuff isn’t being built as fast. The balance between autophagy and protein synthesis is more or less still maintained, that’s why there is no obvious muscle wasting (which can occur when the balance is tilted towards either side[source]), it’s just that both of them are less. This could explain the largely lower levels of many metabolites seen in Naviaux’s study, as a lot of them are to some degree controlled by mTOR.
But perhaps more importantly, this metabolic stagnation would mean that after exercise there is a lot of damaged muscle cells that are not being repaired fast enough. This would, in turn, alarm the immune system[source], possibly contributing to increased autoantibody production, and creating another, this time systemic, feedback loop. It is this loop, along with the ones I mentioned at the start (CGRP, lower myokines) that I believe constitutes Post Exertional Malaise, as PEM is a systemic and delayed reaction to exercise.
Now, going back a little, to the downregulated phosphatidylcholine production. I believe that there are other factors contributing to this, aside from just the Kennedy pathway responding to the energy deficiency. One such factor could be alteration in gut microbiota, which we know happens in ME/CFS. There is some research in mice, which established that changes in the gut bacterial flora influence levels of phosphatidylcholine detectable in the blood (and so probably, it also means lower production of it in the cells).[source1][source2] Unfortunately, we don’t know how exactly that happens. One paper hypothesized that perhaps some bacteria use up a lot of choline from the ingested food, leaving less available for the host; but in the end we just don’t know. Regardless, it would make a lot of sense with this hypothesis, that perhaps the autoimmunity also contributes to eradication of some helpful gut bacteria, allowing other bacteria to proliferate more, which somehow contributes to lower phosphatidylcholine synthesis in the muscle.
Another thing that I should mention, is the fact that abnormally low phosphatidylcholine levels should be sensed by the transcription factor SRBP1, which would then upregulate production.[source1][source2] How exactly that mechanism works is unclear, so we can only speculate why it doesn’t happen in ME/CFS patients. Maybe the mechanism goes through mTOR[source1?][source2?] and can’t activate because there is already a shortage in phosphatidic acid? Maybe the AMP influence over choline kinase overrides SRBP1’s? Maybe because of the dysbiosis choline is the limiting factor? Or maybe ME/CFS patients have some mutation or other mechanism limiting SRPB1’s activity. I don’t know.
I want to also touch a bit on the issue of other cell types. Metabolomics data show us that a metabolic shift is occurring in patients, but they don’t show us where, in what tissue, what cell types. They measure levels of metabolites in the blood, and the idea is that if some stuff is being made less in the cells, then less of it will find its way into the bloodstream. The reason why I focused on skeletal muscle here, is because it is the body’s dominant metabolic site, and because the hallmark symptom of ME/CFS is bad reaction to physical exertion (skeletal muscle contraction). However, it could be the case that the metabolic shift visible in the blood results occurs somewhere else entirely in the body. And even if I am right with my hypothesis here on skeletal muscle, I imagine a similar metabolic shift would have to also occur somewhere in the brain, because mental exertion or sensory overload can also cause PEM (albeit its a bit different from the physical one). Neurons don’t have beta adrenergic receptors, but they do have muscarinic ones, the antibodies to which were also discovered by Scheibenbogen’s group.[source] And microglia have beta adrenergic receptors, which seem to contribute to their activation.[source] There also have been studies on rats showing cAMP efflux and conversion to adenosine in cerebral cortex.[source] I might write a follow-up piece on this in the future, if I connect enough dots.
Additionally, a similar shift in various blood cells could result in easier blood clotting[source] and may potentially explain the “sticky blood” phenomenon in ME/CFS that was talked about recently. (Although probably not in platelets themselves - here a dysfunction in AMPK could inhibit blood clotting instead[source])
Finally, I want to clarify that while I centered this hypothesis around beta-adrenergic antibodies, the same thing could be caused by other types of autoantibodies. For instance, to go all the way back to my previous big hypothesis, an antibody to CD73, which is responsible for the breakdown of extracellular AMP (from cAMP) to adenosine, would have the same effect. Or, more likely, an antibody to the not-yet-classified ecto-phosphodiesterase, which converts extracellular cAMP to AMP. The most that we know about this protein, assuming that there is only one, is that it shares homology with PDE8.[source] Just out of curiosity, I checked PDE8’s sequence against the short sequence identified in Lombardi’s random peptide study. And it matches half of it, that is, three amino acids in a row. It has LSG from GVALSG. I don’t know if that would make for any meaningful binding, but perhaps the actual ecto-phosphodiesterase contains the whole sequence, not just half.
Addendum 1 - Visual diagram of this hypothesis
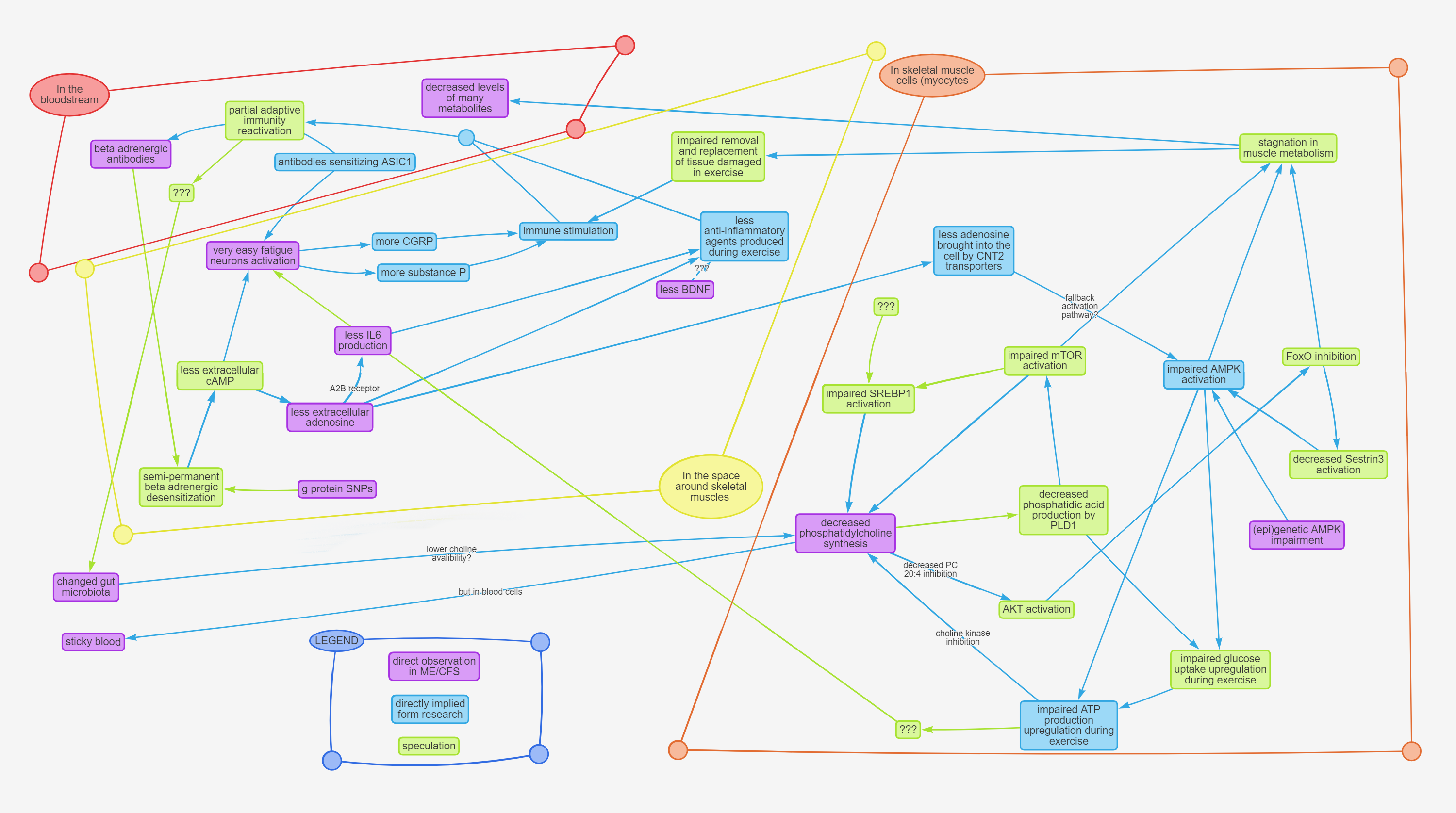
Imgur mirror: https://i.imgur.com/5hH2YwE.png
Addendum 2 - Visual diagram of metabolic signaling pathways in a skeletal muscle myotube.
This diagram is very incomplete, lacking spatial and temporal context, and some of the connections are unclear because of conflicting data, but it’s the best I could manage to make for the purposes of this hypothesis, based on the papers I found.
This is a very large image, so I uploaded it to a hosting site with comfortable zooming and panning: https://www.zoomo.ca/image/ML8H
If you wish to download the original, here is an imgur mirror: https://i.imgur.com/71xhJYF.png
Not portrayed: the full relationship between the Ras/ERK pathway and the PI3K/Akt/mTOR pathway, because there isn’t enough space for an appropriate number of question marks.[source]
Addendum 3 - Treatment
Just because I know people are gonna ask this - if this hypothesis turns out to be correct, what would the treatment be? Frankly, I don’t know. The intertwining of feedback loops here is kinda hard to untangle and it would require solid data, careful planning, and some clever ideas. It also largely depends on which version of this hypothesis would be correct, and to what degree. It is probably not as simple as just taking drugs that activate mTOR or AMPK, because the problem is, that these things are dynamic, and have to be dynamic, adjusting their activation level to the needs of each individual cell. I think perhaps the reason why some people have improvement after taking drugs which activate those things (ketamine, T3), but then that improvement quickly disappears, is because this activation will always be mediated by upregulation of Phospholipase D, which provides more phosphatidic acid for a short period, but further exhausts the phosphatidylcholine stores of the cell. I know that some patients have long term improvement from rapamycin, and the way I can make sense of it, is that by inhibiting mTOR at rest, it gives AMPK a little more breathing room (cause mTOR inhibits AMPK), so slightly more phosphatidylcholine is being made, which allows AMPK more effect over glucose uptake, and, paradoxically, makes it easier for mTOR to activate when it really needs to, after exercise. In the light of this, rapamycin might not be a bad idea, but it’s certainly not a cure. Other than that, perhaps intravenous adenosine and choline could offer some relief, but don’t take my word for it.
Afterword
Thanks for your time. I will be answering a couple rounds of questions, but I will skip any questions that are already answered in the text above. I hope you can forgive me this - if I had enough energy, believe me, I would answer all of them. And please don’t quote this entire post in yours when you’re replying in this thread, it makes a real mess of things.
Credit to @Murph for pointing out to me that adenosine is super low in the metabolomics results , and to Alan Light for his insights into fatigue neurons.
Also tagging @nandixon @Hip @JaimeS because I want to hear their opinion on this.
(and yes, the thing has already been sent to Ron and his team)
Sadly, I have to inform you that I've found a critical flaw in this hypothesis. There are still many pieces to it which I would consider valuable ideas, but the whole no longer works together.
I have stated that "after activating the A1 receptors, a significant portion of this adenosine is actually brought into the cell by CNT2 transporters, and proceeds to activate AMPK. The exact mechanism by which it accomplishes that is not known, but my speculation is that it’s a different mechanism from the one AMP uses, and therefore serves as a fallback activation route in muscle exertion." - this is not fully correct. The adenosine is transported into the cells, and does activate AMPK, but the mechanism is not unknown. The adenosine turns into AMP, which activates AMPK in the usual manner. Meaning, this cannot be an alternative, fallback activation route for AMPK in contracting skeletal muscle. Unfortunately, this is one of the very few places where an error like this means the whole hypothesis falls apart. The two halves of this hypothesis - the systemic part with the anti-beta2 antibodies, and the intracellular part, with the phosphatidic acid - no longer fit together, as they were tied by this idea of adenosine constituting a fallback activation route.
This is fully my mistake, I missed an important part in one of my sources. I apologize terribly for wasting your time. The irony of working on this for half a year only to discover a critical flaw days after going public, would be funny to me if I wasn't so embarrassed by this.
I am leaving this post here, but the untrue statements as well as my ideas directly stemming from this misunderstanding, will be marked red.
The Adenosine - Phosphatidic Acid Hypothesis
Foreword
Hello everyone, my name is Matt, you may remember me as the guy from the CD39 hypothesis (if not: Part1, Part2). Today, I want to present you with something better, the result of my ponderings since that time. It attempts to tie together metabolism, autoimmunity, gut dysbiosis, AMPK, mTOR, ATP production, and explain all the major symptoms, as well as the metabolomics results. I call it the Adenosine - Phosphatidic Acid hypothesis, or the Adn-PA hypothesis for brevity.
Before you start reading, I have to warn you of two things. First, I decided that I will not include explanations for anything that you can easily google by yourself (eg what is AMPK and what it does), because honestly I had no energy left for that. And second, this hypothesis is complex. It is comprised of a dozen hypothetical feedback loops that all intertwine and interact with each other. If you wish to fully understand it, I would recommend reading while looking at the at the diagrams provided in Addendum 1 and Addendum 2, following the pathways in your mind, and perhaps also re-reading it a few times.
If you wish to see a summary first, before reading the whole thing, the diagram in Addendum 1 can serve as such.
The thing itself
Let’s start with the autoantibodies to beta-2 adrenergic receptors. They were discovered in about 30% of ME/CFS patients by Carmen Shcheibenbogen[source], and subsequently confirmed by other researchers. In this theory I am assuming that the rest 70% of patients have other yet undiscovered autoantibodies that achieve a similar effect, or that the T cells are doing a similar thing (both of which have been hinted at by researchers), or perhaps that the effects I describe below have also other systemic mediators. Some ideas for that I will include at the end.
The origin of those antibodies, I imagine, is genetic predisposition for autoimmunity, coupled with a triggering event which flipped some key epigenetic switch, allowing it to first take place. We know that, for example, the Epstain-Barr virus can do this.[source] The reason for why those antibodies persist, and even increase with response to exercise (my speculation), as well as how they cause ME/CFS symptoms, will be the subject of this hypothesis.
Beta-2-ARs are one of the most prominent G protein coupled receptors. They couple mainly with Gs types, meaning they stimulate adenylyl cyclase to produce cAMP, which then activates Protein Kinase A.[source] This action can sometimes get very difficult to track, because its effects don’t just differ between cell types, they also differ between specific localization in the cell. Cells with G protein coupled receptors have individual “pockets” inside them with a specific receptor, a localized pool of cAMP, phosphodiesterases surrounding it, preventing from influencing other pockets, and localized Protein Kinase A attached to a specific AKAP (A Kinase Anchoring Protein). There is about 40-something types of those, and they strongly modulate what the attached Protein Kinase A does.[source1][source2][source3][source4] So in one single cell, a G protein coupled receptor on one end of the cell, and a second receptor on the other end of the cell, could have very different functions, despite being the exact same type of a receptor, because their PKAs are attached to different AKAPs and their cAMP pools are separated by PDEs.
Fortunately, there are some aspects that are common. Of particular interest to me, is the mechanism of desensitization of those receptors. When cAMP levels rise, this activates MRP transporters which bring a significant portion of the cAMP outside of the cell. The cAMP is then converted to AMP by a yet-to-be-classified ecto-phosphodiesterase, and then the AMP is turned into adenosine by ecto-nucleotidase CD73. The adenosine then stimulates the A1 receptors of the cell, which are also GPCRs, but couple primarily with Gi types, so they inhibit cAMP production by adenylyl cyclases. This closes the loop of a negative feedback cycle, suppressing cAMP production completely after 15-60 minutes of constant receptor stimulation. This effect has been observed in many CGRPs, including beta adrenergic receptors.[source1][source2][source3]
This mechanism is interesting for a few reasons. Firstly, the autoantibodies in ME/CFS cause gain of function of the beta2 receptors - they make them activate more. That should mean more extracellular adenosine. And yet, looking at the metabolomics data from Robert Naviaux, andenosie is actually one of the most decreased compounds. It is extremely low.[source] There are, obviously, other sources of adenosine in the bloodstream, but my impression is that GPCR desensitization is one of the largest ones, if not the dominant one. This leads me to believe, that what happens in ME/CFS patients, is the cells after prolonged constant stimulation of their beta adrenergic receptors, take compensative measures to offset this. Perhaps by suppressing the genetic expression of those receptors, or G proteins, or by uncoupling them somehow. I don’t know how exactly. But the effect is a diminished and very constant level of activation, that is not very responsive to the proper highs and lows of the actual ligand concentration. In the context of vascular smooth muscles, this can explain POTS and orthostatic intolerance very well, as it would mean the blood vessels can’t adapt their width to keep the blood pressure constant, when faced with changing physical conditions (like standing upright).
But I am more interested in what this does in skeletal muscle. Now, in skeletal muscle the beta adrenergic receptors have some role in potentiation of the muscle contractile force, so this might mean some limited muscle weakness, which does seem to happen to patients. But I believe that a much more important change occurs because of the diminished extracellular adenosine production, as the cAMP levels never rise very high. In healthy people, the activation of skeletal muscle beta adrenergic receptors, and the subsequent adenosine production would presumably occur mainly in exercise, as that is when blood noradrenaline levels are high, and when noradrenaline is released from surrounding neurons.[source1][source2] There are even studies directly showing higher adenosine production after exercise.[source] So in my hypothesis here, the patients have lower resting adenosine levels, but much more importantly, those levels do not rise as they would in a healthy person during exercise. And I believe I have solid grounds to suspect that this rise in extracellular adenosine has multiple important roles during muscle exertion, and is not simply a side product of GPCR desensitization.
First such role is anti-inflammatory. It has been observed in multiple immune cell types, such as dendritic cells and macrophages, that extracellular adenosine attenuates their activation, and has even been hypothesized to mediate the mechanism of their deactivation in immune resolution.[source1][source2][source3][source4] This is important because the immune system participates to some degree in the muscle repair following exercise.[source1][source2][source3][source4][source5] And my guess is that adenosine serves as a moderator, keeping the immune cells in check and preventing them from reacting in an autoimmune manner.
Another such moderator is probably IL6, the interleukin that is also produced by muscle during exercise.[source] And in many cell types it has been observed that activation of the adenosine receptor A2B elicits IL6 production and secretion.[source][source2][source3][source4] It’s not a far-fetched idea that the same would happen in muscle cells. And so, if adenosine is lower, IL6 is lower.
(Another such factor might be BDNF[source], which is sometimes found to be low in ME/CFS as well, but I haven’t explored this enough to write about it yet)
Without those immune moderators, which are normally present during exercise, ME/CFS patients’ immune systems, activated by the muscle exertion, are more likely to go overboard, and autoimmune. And proceed to produce even more anti-beta2 antibodies and/or T cells, closing the disease feedback loop. The first one at least. There is more.
The feeling of fatigue is facilitated by synergistic activation of ASIC, TRPV1 and P2X3 receptors on certain nociceptive neurons. The main receptor is ASIC, with TRPV1 and P2X3 working to sensitize it, to activate more easily. The metabolites required for this activation are lactic acid and extracellular ATP (both of which are released from muscle cells during exercise).[source1][source2] And it just so happens that extracellular cAMP attenuates this process.[source] Meaning, if those neurons activate in healthy people in presence of lots of extracellular cAMP, and that gets you normal fatigue, then it makes sense than in ME/CFS patients, with much lower cAMP efflux, they activate more strongly and easily, resulting in intense fatigue. Certain levels of LA and ATP are present there even at rest, so without the inhibitory action of cAMP they could very well be activating even at rest.
But I think there is yet another mechanism at play here. There is a study that I keep going back to, done by Vincent Lombardi’s team, where they tested antibodies in serum of ME/CFS patients against 125000 peptides with random amino acid sequences, to find what the antibodies are reacting to, even if it’s a new unknown antibody. The study didn’t find a very specific match, but they did find a short sequence that, if I understand correctly, antibodies in the great majority of patients react to. And after they did a search to find human proteins containing it, or a similar sequence, among the 30 best matches for it was the ASIC1 receptor.[source] The main receptor responsible for facilitating the feeling of fatigue. Personally, I find it unlikely for this to be a coincidence, so what I hypothesize is happening here, is that antibodies with this sequence bind to the ASIC receptors and sensitize them in a similar manner that eg. the P2X3 receptor does. This acts synergistically with the lower levels of extracellular cAMP to activate the fatigue neurons more easily and strongly.
I’m writing about this not only to explain why the patients have intense fatigue, but also because this can very well constitute another feedback cycle reinforcing the disease. As Alan Light pointed out to me, the nociceptive neurons, when activating, secrete certain peptides like CGRP[source], and Substance P[source], which can modulate the behaviour of immune cells.[source] Hence it is possible that the activation of fatigue neurons itself also reinforces the autoimmunity.
All of the above by itself constitutes a hypothetical disease mechanism that explains a lot of the symptoms, but what I really wanted this to do, is to explain the metabolomics results, which I still consider the most important data we currently have on ME/CFS.
To do that, I must first talk about Julia Newton’s research regarding AMPK. Her team demonstrated that in skeletal muscle cells cultured from ME/CFS patients, and stimulated for contraction, there is less AMPK activity than in cells from healthy subjects.[source] I find this discovery particularly important precisely because it is in skeletal muscle cells (myotubes), and not in PBMCs (I explained why here). I’ve come up with dozens of ways for an autoimmune mechanism to cause AMPK impairment in skeletal muscle, but none of them can explain these results for one reasons - the cells in this study were not myotubes taken directly from patients, they were cultivated in a dish from patients’ muscle satellite cells, which are kind of resident blueprints for myotubes, used to expand and repair them when need be. What this means is that the myotubes in the dish share only the genetic and epigenetic factors, and not other influences, with the myotubes in the patients. So all of the processes that I described above, even if present in patients’ skeletal muscles, would not translate to the muscle cells in the dish here.
Therefore, what I propose, is that the impairments in AMPK activation in skeletal muscle which were identified in this study, are strictly genetic or epigenetic, and that patients carry them their whole life, even before developing ME/CFS. This impairment itself is not enough to cause the disease, but it lies very important groundwork for it to take place.
The reason why I think this, is because I believe there exist fallback mechanisms that make sure AMPK is activated during skeletal muscle contraction, which are independent of the direct activation by AMP accumulating in the cell. Particularly, I think I’ve found one such mechanism, which relies on the extracellular adenosine created in muscle contraction. As it turns out, after activating the A1 receptors, a significant portion of this adenosine is actually brought into the cell by CNT2 transporters, and proceeds to activate AMPK.[source] The exact mechanism by which it accomplishes that is not known, but my speculation is that it’s a different mechanism from the one AMP uses, and therefore serves as a fallback activation route in muscle exertion.
So, I propose that people with (epi)genetic impairment of AMPK activation in skeletal muscle stay healthy and don’t develop ME/CFS, because they still have this fallback mechanism working. They only get ME/CFS after developing autoimmunity to beta adrenergic receptors, which leads to impaired adenosine production in the extracellular space surrounding skeletal muscle, and leads to loss of this fallback mechanism. The result is AMPK that doesn’t sufficiently activate when the muscles become active, leading to uptake of glucose and ATP synthesis not becoming appropriately upregulated during exercise. The Krebs cycle, glycolysis and oxidative phosphorylation have other regulation mechanisms independent of AMPK, so this probably isn’t enough of an impairment for cells to die, but is enough to elicit some changes in them.
What changes exactly? Lots of things, probably; most of which I don’t know about. It could also perhaps activate the fatigue neurons further - I am skeptical of this idea because I haven’t seen any research demonstrating that this is possible, but I’ve seen a lot of researchers hint at this, and it certainly would make a lot of evolutionary sense for the fatigue neurons to be able to react directly to the energy state of the muscles, so I’m keeping an open mind. (If you know of any research demonstrating such a pathway existing, pretty please send it my way)
But here I wanted to focus on one particular consequence, which I haven’t seen addressed before, namely the effect on the Kennedy (CDP-choline) pathway. This pathways synthesizes phosphatidylcholine from choline, using ATP for the process. And it becomes largely downregulated if the cell doesn’t have plentiful energy. This is actually fully independent of AMPK - the choline kinase somehow senses the ATP/AMP ratio directly.[source1][source2]
My hypothesis here is that after a long time of the Kennedy pathway being inhibited by the low energy status, the skeletal muscle cells of ME/CFS patients start running low on phosphatidylcholine. This is consistent with the metabolomics results - the main thing that the two large metabolomics studies of Robert Naviaux[source] and Ian Lipkin[source] agree on, is that phosphatidylcholines are decreased.
The main role of phosphatidylcholine is to be a building material for cell membranes, but I wanted to focus on a different role here.
Phospholipase D is an enzyme that breaks down phosphatidylcholine to make phosphatidic acid. And I’ve found some really interesting papers demonstrating the very important role that this reaction plays in the regulation of mTOR and AMPK.
mTOR is a master regulator of protein synthesis in cells, and it is regulated by the TSC1/TSC2 complex, which, when active, prevents Rheb from associating with GTP, and only GTP-loaded Rheb can activate mTOR. All the pathways which influence mTOR go through here, either activating or inhibiting TSC1/TSC2 to control what mTOR is doing. And it turns out that the de facto mechanism by which GTP-loaded Rheb activates mTOR involves Phospholipase D1. This phospholipase is activated by GTP-Rheb, producing phosphatidic acid, which in turn displaces the inhibiting protein FKBP38 from mTOR, allowing Rheb to bind to mTOR in its place and activate it. If Phospholipase D1 action is inhibited, this process cannot occur and mTOR cannot activate.[source1][source2]
Phospholipase D1 also has an equally important role with AMPK. One would think that since mTOR and AMPK have an antagonistic relationship, that Phospholipase D1 activity would inhibit AMPK, or the other way around, and there is indeed one paper claiming that.[source] But there is more and much more thorough evidence, that AMPK actually activates Phospholipase D1. Moreover, the resulting phosphatidic acid, by facilitating translocation of Raf1 to cell membrane, is the mechanism by which AMPK activates the ERK/MAPK cascade and upregulates glucose uptake in times of higher energy demand. (and probably, it’s also the mechanism by which Akt/PKB activates ERK/MAPK, since Akt lies upstream of TSC1/TSC2) If Phospholipase D1 is inhibited, AMPK cannot fully upregulate glucose uptake.[source1][source2][source3]
With all this said, I do not think Phospholipase D1 action is defective in ME/CFS. Rather, because of the aforementioned phosphatidylcholine deprivation, Phospholipase D1 runs out of substrate for breaking down into phosphatidic acid.
If this becomes the case, even if the prior impairment in AMPK activation was fairly minor, the lack of phosphatidic acid production further prevents AMPK action, not by directly inhibiting it, but by making it unable to upregulate glucose uptake properly in times of greater energetic demand, starving the Krebs cycle of fuel, which further inhibits the Kennedy pathway, and we have another feedback loop.
It also would strongly inhibit mTOR activation. But, interestingly enough, it’s possible that Akt would actually be upregulated here. This is because a particular kind of phosphatidylcholine - PC 20:4, is an Akt inhibitor[source], and with its production downregulated, Akt might activate more. This would lead to inhibition of FoxO factors, acting synergistically with the inhibited AMPK (AMPK also activates FoxO). This inhibition of FoxO could lead to downregulation of Sestrin 3, which is another AMPK activator, potentially furthering AMPK inhibition.[source]
But more importantly, FoxO3 is an important mediator of autophagy. While it is not directly stated anywhere in the literature, it is my understanding after reading it, that AMPK and mTOR act synergistically to repair damage done to the muscles during exercise. AMPK acts first, activating FoxO3, enabling autophagy, which removes all the damaged parts.[source1][source2] Then mTOR mediates synthesis of new proteins to replace them.[source1][source2] And elevated IL6 levels signal the satellite cells to help with this process also, providing new nuclei etc for the myotubes undergoing repair.[source]
So what I propose occurs in the muscles of ME/CFS patients, is metabolic stagnation, caused by AMPK and mTOR not activating properly in exercise, and also by IL6 not being secreted as much. Metabolic stagnation, meaning, old stuff isn’t broken down as fast, and new stuff isn’t being built as fast. The balance between autophagy and protein synthesis is more or less still maintained, that’s why there is no obvious muscle wasting (which can occur when the balance is tilted towards either side[source]), it’s just that both of them are less. This could explain the largely lower levels of many metabolites seen in Naviaux’s study, as a lot of them are to some degree controlled by mTOR.
But perhaps more importantly, this metabolic stagnation would mean that after exercise there is a lot of damaged muscle cells that are not being repaired fast enough. This would, in turn, alarm the immune system[source], possibly contributing to increased autoantibody production, and creating another, this time systemic, feedback loop. It is this loop, along with the ones I mentioned at the start (CGRP, lower myokines) that I believe constitutes Post Exertional Malaise, as PEM is a systemic and delayed reaction to exercise.
Now, going back a little, to the downregulated phosphatidylcholine production. I believe that there are other factors contributing to this, aside from just the Kennedy pathway responding to the energy deficiency. One such factor could be alteration in gut microbiota, which we know happens in ME/CFS. There is some research in mice, which established that changes in the gut bacterial flora influence levels of phosphatidylcholine detectable in the blood (and so probably, it also means lower production of it in the cells).[source1][source2] Unfortunately, we don’t know how exactly that happens. One paper hypothesized that perhaps some bacteria use up a lot of choline from the ingested food, leaving less available for the host; but in the end we just don’t know. Regardless, it would make a lot of sense with this hypothesis, that perhaps the autoimmunity also contributes to eradication of some helpful gut bacteria, allowing other bacteria to proliferate more, which somehow contributes to lower phosphatidylcholine synthesis in the muscle.
Another thing that I should mention, is the fact that abnormally low phosphatidylcholine levels should be sensed by the transcription factor SRBP1, which would then upregulate production.[source1][source2] How exactly that mechanism works is unclear, so we can only speculate why it doesn’t happen in ME/CFS patients. Maybe the mechanism goes through mTOR[source1?][source2?] and can’t activate because there is already a shortage in phosphatidic acid? Maybe the AMP influence over choline kinase overrides SRBP1’s? Maybe because of the dysbiosis choline is the limiting factor? Or maybe ME/CFS patients have some mutation or other mechanism limiting SRPB1’s activity. I don’t know.
I want to also touch a bit on the issue of other cell types. Metabolomics data show us that a metabolic shift is occurring in patients, but they don’t show us where, in what tissue, what cell types. They measure levels of metabolites in the blood, and the idea is that if some stuff is being made less in the cells, then less of it will find its way into the bloodstream. The reason why I focused on skeletal muscle here, is because it is the body’s dominant metabolic site, and because the hallmark symptom of ME/CFS is bad reaction to physical exertion (skeletal muscle contraction). However, it could be the case that the metabolic shift visible in the blood results occurs somewhere else entirely in the body. And even if I am right with my hypothesis here on skeletal muscle, I imagine a similar metabolic shift would have to also occur somewhere in the brain, because mental exertion or sensory overload can also cause PEM (albeit its a bit different from the physical one). Neurons don’t have beta adrenergic receptors, but they do have muscarinic ones, the antibodies to which were also discovered by Scheibenbogen’s group.[source] And microglia have beta adrenergic receptors, which seem to contribute to their activation.[source] There also have been studies on rats showing cAMP efflux and conversion to adenosine in cerebral cortex.[source] I might write a follow-up piece on this in the future, if I connect enough dots.
Additionally, a similar shift in various blood cells could result in easier blood clotting[source] and may potentially explain the “sticky blood” phenomenon in ME/CFS that was talked about recently. (Although probably not in platelets themselves - here a dysfunction in AMPK could inhibit blood clotting instead[source])
Finally, I want to clarify that while I centered this hypothesis around beta-adrenergic antibodies, the same thing could be caused by other types of autoantibodies. For instance, to go all the way back to my previous big hypothesis, an antibody to CD73, which is responsible for the breakdown of extracellular AMP (from cAMP) to adenosine, would have the same effect. Or, more likely, an antibody to the not-yet-classified ecto-phosphodiesterase, which converts extracellular cAMP to AMP. The most that we know about this protein, assuming that there is only one, is that it shares homology with PDE8.[source] Just out of curiosity, I checked PDE8’s sequence against the short sequence identified in Lombardi’s random peptide study. And it matches half of it, that is, three amino acids in a row. It has LSG from GVALSG. I don’t know if that would make for any meaningful binding, but perhaps the actual ecto-phosphodiesterase contains the whole sequence, not just half.
Addendum 1 - Visual diagram of this hypothesis
Imgur mirror: https://i.imgur.com/5hH2YwE.png
Addendum 2 - Visual diagram of metabolic signaling pathways in a skeletal muscle myotube.
This diagram is very incomplete, lacking spatial and temporal context, and some of the connections are unclear because of conflicting data, but it’s the best I could manage to make for the purposes of this hypothesis, based on the papers I found.
This is a very large image, so I uploaded it to a hosting site with comfortable zooming and panning: https://www.zoomo.ca/image/ML8H
If you wish to download the original, here is an imgur mirror: https://i.imgur.com/71xhJYF.png
Not portrayed: the full relationship between the Ras/ERK pathway and the PI3K/Akt/mTOR pathway, because there isn’t enough space for an appropriate number of question marks.[source]
Addendum 3 - Treatment
Just because I know people are gonna ask this - if this hypothesis turns out to be correct, what would the treatment be? Frankly, I don’t know. The intertwining of feedback loops here is kinda hard to untangle and it would require solid data, careful planning, and some clever ideas. It also largely depends on which version of this hypothesis would be correct, and to what degree. It is probably not as simple as just taking drugs that activate mTOR or AMPK, because the problem is, that these things are dynamic, and have to be dynamic, adjusting their activation level to the needs of each individual cell. I think perhaps the reason why some people have improvement after taking drugs which activate those things (ketamine, T3), but then that improvement quickly disappears, is because this activation will always be mediated by upregulation of Phospholipase D, which provides more phosphatidic acid for a short period, but further exhausts the phosphatidylcholine stores of the cell. I know that some patients have long term improvement from rapamycin, and the way I can make sense of it, is that by inhibiting mTOR at rest, it gives AMPK a little more breathing room (cause mTOR inhibits AMPK), so slightly more phosphatidylcholine is being made, which allows AMPK more effect over glucose uptake, and, paradoxically, makes it easier for mTOR to activate when it really needs to, after exercise. In the light of this, rapamycin might not be a bad idea, but it’s certainly not a cure. Other than that, perhaps intravenous adenosine and choline could offer some relief, but don’t take my word for it.
Afterword
Thanks for your time. I will be answering a couple rounds of questions, but I will skip any questions that are already answered in the text above. I hope you can forgive me this - if I had enough energy, believe me, I would answer all of them. And please don’t quote this entire post in yours when you’re replying in this thread, it makes a real mess of things.
Credit to @Murph for pointing out to me that adenosine is super low in the metabolomics results , and to Alan Light for his insights into fatigue neurons.
Also tagging @nandixon @Hip @JaimeS because I want to hear their opinion on this.
(and yes, the thing has already been sent to Ron and his team)
Last edited:
")