I know that some amount of Methyl B12 converts to Adeno B12, but can the reverse happen also? You don't have to get into too many of the details, as a simply yes or no would suffice, providing you know what you're talking about. I would also like people to stick to the topic (not get into the importance of each, etc.), thanks.

-

Welcome to Phoenix Rising!
Created in 2008, Phoenix Rising is the largest and oldest forum dedicated to furthering the understanding of, and finding treatments for, complex chronic illnesses such as chronic fatigue syndrome (ME/CFS), fibromyalgia, long COVID, postural orthostatic tachycardia syndrome (POTS), mast cell activation syndrome (MCAS), and allied diseases.
To become a member, simply click the Register button at the top right.
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
Can Adeno B12 convert to Methyl B12?
- Thread starter NC360
- Start date
ahmo
Senior Member
- Messages
- 4,805
- Location
- Northcoast NSW, Australia
no. 2 separate things.
but can the reverse happen also
Yes. The adenosyl form is used for storage in the liver. It is converted to the methyl form as needed.
no. 2 separate things.
Yes. The adenosyl form is used for storage in the liver. It is converted to the methyl form as needed.
Two different answers? Which is correct?

Two different answers? Which is correct?
Here is a recent review which covers some of the issues.
Here are some quotes
The primary storage sites for Cbl are the liver and kidney. The liver contains ∼10 μg Cbl/g protein or 1 μg/g wet weight and can carry between 1–1·5 mg of the vitamin. The stored amount increases with age from newborn to adult (Rappazzo et al, 1970). As the bulk of the liver mass is due to hepatocytes, HC-bound Cbl taken up via the asialoglycoprotein receptor could account for most of the stored Cbl. (Alpers & Russell-Jones, 1999).
The intracellular distribution of various forms of Cbl has been well documented and in all tissues 5′-deoxyadenosyl Cbl (AdoCbl) is the predominant form with lesser amounts of hydroxo-Cbl. Methyl Cbl (MeCbl) is a minor component of intracellular Cbl. However, MeCbl is the major form of Cbl in the plasma and it is this form that is disproportionately reduced in Cbl deficiency.
The initial steps in the synthesis of Cbl coenzymes appear to be in the efficient removal of the upper axial ligand attached to the central cobalt, irrespective of the form of Cbl transported into the cell as shown by efficient and rapid interconversion of labelled cyano-Cbl, AdoCbl and MeCbl (Quadros et al, 1979).
I thought I had another reference but can't find it at the moment.
If you need more detail you'll have to search for it yourself.
Does anyone know if Hydroxy B12 converts better to Methyl B12 when compared to Adeno B12?
There is no reason to think it would be though I haven't seen any figures. The study I referenced above showed that that there was rapid interconversion of cyano, methyl and adenosyl forms (hydroxyl wasn't mentioned).
Since the adenosyl form is the major storage form and methyl is the major form in blood, the most common interconversion in the body would be between these forms.
I'm pretty sure hydroxoB12 is the storage form. And wikipedia says that adenosylB12 and methylB12 can't convert to each other:Since the adenosyl form is the major storage form and methyl is the major form in blood, the most common interconversion in the body would be between these forms.
Methylcobalamin is not sufficient as a singular source of vitamin B12. Hydroxocobalamin and cyanocobalamin can both be split by the body into methylcobalamin and adenosylcobalamin. Methylcobalamin on the other hand is not converted into adenosylcobalamin. Deficiency of adenosylcobalamin disturbs carbohydrate, fat and amino-acid metabolism, and hence interferes with the formation of myelin.[3] Thereby it is important to treat vitamin B12 deficiency with hydroxocobalamin or cyanocobalamin or a combination of adenosylcobalamin and methylcobalamin.[3]
I'm pretty sure hydroxoB12 is the storage form. And wikipedia says that adenosylB12 and methylB12 can't convert to each other:
I'm quoting the paper I referenced above and I read several other sources saying that adenosyl is the major storage form.
I've tried to find more info on interconversion previously but without much success. In the study cited, cyano, methyl and adenosyl interconverted readily.
Most of the literature on this seems to be very old and not readily available so I've not been able to find a lot of info.
Is there any info on which enzymes do the converting? That would help us figure it outI've tried to find more info on interconversion previously but without much success. In the study cited, cyano, methyl and adenosyl interconverted readily.
Is there any info on which enzymes do the converting?
The review talks about this a bit but I get the impression it is complicated and not fully understood. They make the statement
A system for efficient reduction of cobalt in Cbl and interconversion of all forms of Cbl probably involves multiple enzymes and cofactors
Delving a bit more into the Cbl letter diseases might give a bit more info (they produce a table about this).
I'll search again tomorrow and try to follow some of the references from the review but I'm too tired now.
I bought a new car today - very satisfying but also exhausting.
This is something I have been meaning to track down for some time - I should just go to the local medical school library - maybe later.
Many sites claim that the two forms can be converted to each other but don't give details.
Ben Lynch said here: "Maybe they have a genetic defect or some enzymatic co-factor problem that converts methylcobalamin to adenosylcobalamin. That conversion occurs in the mitochondria, because adenosylcobalamin is a mitochondrial form of B12."
VeganHealth cited a report as follows:
Ben Lynch said here: "Maybe they have a genetic defect or some enzymatic co-factor problem that converts methylcobalamin to adenosylcobalamin. That conversion occurs in the mitochondria, because adenosylcobalamin is a mitochondrial form of B12."
VeganHealth cited a report as follows:
Donaldson (2000, USA) studied 3 vegans with elevated urinary MMA levels who were treated with 1/2 to 1 sublingual MeCbl tablet, 2 times/day for 3 weeks. Correspondence with the author (March 21, 2002) verified that these tablets contained 1,000 µg MeCbl each.
Two of the subjects' urinary MMA normalized while the remaining subject's stayed slightly elevated at 4.1 µg/mg creatinine (normal is < 4.0 µg/mg creatinine). Thus, at a rate of 1,000-2,000 µg/day, MeCbl appears to be absorbed at a high enough rate to improve B12 status in some vegans. Additionally, this indicates that the MeCbl was converted to AdoCbl for use in the MMA pathway.
Two of the subjects' urinary MMA normalized while the remaining subject's stayed slightly elevated at 4.1 µg/mg creatinine (normal is < 4.0 µg/mg creatinine). Thus, at a rate of 1,000-2,000 µg/day, MeCbl appears to be absorbed at a high enough rate to improve B12 status in some vegans. Additionally, this indicates that the MeCbl was converted to AdoCbl for use in the MMA pathway.
I just remembered this paper:
Processing of alkylcobalamins in mammalian cells: a role for the MMACHC (cblC) gene product
Processing of alkylcobalamins in mammalian cells: a role for the MMACHC (cblC) gene product
Abstract
The MMACHC gene product of the cblC complementation group, referred to as the cblC protein, catalyzes the in vitro and in vivo decyanation of cyanocobalamin (vitamin B12). We hypothesized that the cblC protein would also catalyze the dealkylation of newly internalized methylcobalamin (MeCbl) and 5′-deoxyadenosylcobalamin (AdoCbl), the naturally occurring alkylcobalamins that are present in the diet. The hypothesis was tested in cultured endothelial cells using [57Co]-AdoCbl and MeCbl analogs consisting of [57Co]-labeled straight-chain alkylcobalamins ranging from C2 (ethylcobalamin) to C6 (hexylcobalamin). [57Co]-AdoCbl was converted to [57Co]-MeCbl by cultured bovine aortic endothelial cells, suggesting that a dealkylation process likely involving the cblC protein removed the 5′-deoxyadenosyl alkyl group. Surprisingly, all of the straight-chain alkylcobalamins served as substrates for the biosynthesis of both AdoCbl and MeCbl. Dealkylation was then assessed in normal skin fibroblasts and fibroblasts derived from 3 patients with mutations in the MMACHC gene. While normal skin fibroblasts readily converted [57Co]-propylcobalamin to [57Co]-AdoCbl and [57Co]-MeCbl, there was little or no conversion in cblC mutant fibroblasts. These studies suggest that the CblC protein is responsible for early processing of both CNCbl (decyanation) and alkylcobalamins (dealkylation) in mammalian cells.
Many sites claim that the two forms can be converted to each other but don't give details.
That's been my experience as well.
VeganHealth cited a report as follows:
Donaldson (2000, USA) studied 3 vegans with elevated urinary MMA levels who were treated with 1/2 to 1 sublingual MeCbl tablet, 2 times/day for 3 weeks. Correspondence with the author (March 21, 2002) verified that these tablets contained 1,000 µg MeCbl each.
Two of the subjects' urinary MMA normalized while the remaining subject's stayed slightly elevated at 4.1 µg/mg creatinine (normal is < 4.0 µg/mg creatinine). Thus, at a rate of 1,000-2,000 µg/day, MeCbl appears to be absorbed at a high enough rate to improve B12 status in some vegans. Additionally, this indicates that the MeCbl was converted to AdoCbl for use in the MMA pathway.
In some vegans? Or rather those vegans may have gotten it elsewhere somehow? As of now—without good evidence—I will assume they (MeCbl and AdoCbl) don't covert to each other, or rather just AdoCbl converts to MeCbl?
Last edited:
PeterPositive
Senior Member
- Messages
- 1,426
I think you may be right as I was able to deal with high homocysteine with folate + AdoCbl. If it did not convert to methyl-B12 it would have done nothing.In some vegans? Or rather those vegans may have gotten it elsewhere somehow? As of now—without good evidence—I will assume they (MeCbl and AdoCbl) don't covert to each other, or rather just AdoCbl converts to MeCbl?
On the other hand when I added MethylCbl the homocysteine level went down further, which suggests only a part of the AdoCbl had been converted... ?
Just to clarify, there is not direct interconversion of adenosyl and methyl forms, rather all forms of cobalamin transported into the cell have the upper axial ligand removed (ie the methyl, adenosyl, hydroxyl or cyano residue). The cobalamin moiety is then directed to formation of methyl or adenosyl forms as required.
Studies with labelled forms with different upper axial ligands taken up by the cell show that the different forms interconvert readily.
The reverse process in the liver whereby adenosyl is removed from stores and converted to methyl is not understood, but is presumed to take place in several steps.
Studies with labelled forms with different upper axial ligands taken up by the cell show that the different forms interconvert readily.
The reverse process in the liver whereby adenosyl is removed from stores and converted to methyl is not understood, but is presumed to take place in several steps.
I have reread the Quadros review of cobalamin assimilation and metabolism plus a two reviews on the cobalamin letter diseases (inborn errors of metabolism) which help to define the steps in intracellular cobalamin processing.
I was reminded all over again of my original frustration in trying to understand the detail better - every interesting reference in the Quadros review is behind a significant pay wall.
I found out though that Quadros is very active in cobalamin research and has made significant contributions to understanding. So until evidence is produced to the contrary, I'll accept the statements made in the review even if I can't read the detail behind them.
The reviews of the letter diseases (described as cblA - cblG) support what he says.
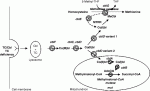
To summarise :-
Cobalamin is stored in liver and kidney. The storage form is not specifically addressed in the review but a more general statement is made that the predominant form of cobalamin in tissues is adenosyl with a small amount of hydroxyl; the form in blood is predominantly methyl. I have seen other studies stating that the storage form is adenosyl. So it seems that adenosyl is the storage form but I would like more direct evidence for this. In any case, the details of storage and release from storage are not understood.
Cobalamin in blood (predominantly the methyl form) is taken into cells bound to the carrier protein transcobalamin 2 (TCN2), via a specific receptor (Quadros was involved in isolation and characterisation of both the carrier protein and the receptor).
The 3-component complex is processed inside the cell with the receptor recycled to the cell surface and cobalamin/TCN2 sent to lysozomes to release the cobalamin from the binding protein.
This is when the first of the errors occurs - (cblF) - cobalamin largely remains stuck in the lysozome.
Normally though free cobalamin with its upper axial ligand attached (ie methyl, adenosyl etc) is released from the lysozome and two steps follow.
The first removes the ligand (the dealkylation reaction described in the reference linked by @Eastman, which is performed by the gene product of MMACHC, defined by cblC.
The second, the product of MMADHC definied by cblD, changes the oxidation state of the cobalt, a cobalamin reductase.
From here, cobalamin is shepherded either to the enzyme complex MTR/MTRR where cobalt is further reduced and a methyl group added, or into the mitochondrion. cblE and G define defects in MTRR and MTR respectively.
Two different variants defined by cblD (variant 1 and 2) appear to act as chaperones in this process, protecting and stabilising cobalamin (ie the cobalamin is never left naked). Variant 1 is involved with MTR/MTRR and variant 2 with MUT.
In the mitochondrion, the product of MMMA, defined by cblA, of uncertain function, but presumably a cobalamin reductase, further reduces the cobalt atom. Then an adenosyltransferase, the product of MMMB, defined by cblB, adds an adenosyl group (in an ATP dependant reaction) before the cofactor binds to its enzyme MUT (methyl malonylcoA mutase).
A defect in MUT is described as MUT rather than a cbl letter.
I have uploaded a diagram.
The other relevant studies were also performed by Quadros. In these, various labelled forms of cobalamin were added to cells and the products analysed. These showed that the three forms studied, viz cyano, adenosyl and methyl, were readily interconverted.
That's all I have the energy for now.
Intracellular processing of cobalamin is very complex and is not fully understood.
Initially though the upper axial ligand is removed from all forms once they are inside the cell. From there, cobalamin can be converted to either of the active forms.
Labelling studies show that the forms can interconvert readily.
Probably adenosyl is the storage form but storage mechanisms are understood even less well.
I was reminded all over again of my original frustration in trying to understand the detail better - every interesting reference in the Quadros review is behind a significant pay wall.
I found out though that Quadros is very active in cobalamin research and has made significant contributions to understanding. So until evidence is produced to the contrary, I'll accept the statements made in the review even if I can't read the detail behind them.
The reviews of the letter diseases (described as cblA - cblG) support what he says.
To summarise :-
Cobalamin is stored in liver and kidney. The storage form is not specifically addressed in the review but a more general statement is made that the predominant form of cobalamin in tissues is adenosyl with a small amount of hydroxyl; the form in blood is predominantly methyl. I have seen other studies stating that the storage form is adenosyl. So it seems that adenosyl is the storage form but I would like more direct evidence for this. In any case, the details of storage and release from storage are not understood.
Cobalamin in blood (predominantly the methyl form) is taken into cells bound to the carrier protein transcobalamin 2 (TCN2), via a specific receptor (Quadros was involved in isolation and characterisation of both the carrier protein and the receptor).
The 3-component complex is processed inside the cell with the receptor recycled to the cell surface and cobalamin/TCN2 sent to lysozomes to release the cobalamin from the binding protein.
This is when the first of the errors occurs - (cblF) - cobalamin largely remains stuck in the lysozome.
Normally though free cobalamin with its upper axial ligand attached (ie methyl, adenosyl etc) is released from the lysozome and two steps follow.
The first removes the ligand (the dealkylation reaction described in the reference linked by @Eastman, which is performed by the gene product of MMACHC, defined by cblC.
The second, the product of MMADHC definied by cblD, changes the oxidation state of the cobalt, a cobalamin reductase.
From here, cobalamin is shepherded either to the enzyme complex MTR/MTRR where cobalt is further reduced and a methyl group added, or into the mitochondrion. cblE and G define defects in MTRR and MTR respectively.
Two different variants defined by cblD (variant 1 and 2) appear to act as chaperones in this process, protecting and stabilising cobalamin (ie the cobalamin is never left naked). Variant 1 is involved with MTR/MTRR and variant 2 with MUT.
In the mitochondrion, the product of MMMA, defined by cblA, of uncertain function, but presumably a cobalamin reductase, further reduces the cobalt atom. Then an adenosyltransferase, the product of MMMB, defined by cblB, adds an adenosyl group (in an ATP dependant reaction) before the cofactor binds to its enzyme MUT (methyl malonylcoA mutase).
A defect in MUT is described as MUT rather than a cbl letter.
I have uploaded a diagram.
The other relevant studies were also performed by Quadros. In these, various labelled forms of cobalamin were added to cells and the products analysed. These showed that the three forms studied, viz cyano, adenosyl and methyl, were readily interconverted.
That's all I have the energy for now.
Intracellular processing of cobalamin is very complex and is not fully understood.
Initially though the upper axial ligand is removed from all forms once they are inside the cell. From there, cobalamin can be converted to either of the active forms.
Labelling studies show that the forms can interconvert readily.
Probably adenosyl is the storage form but storage mechanisms are understood even less well.
Attachments
Last edited:
I thought afterwards that we should consider that cobalamin can also get into cells from blood by passive diffusion - ie independent of the receptor mechanism described above.
This might be particularly relevant when supplementing relatively large amounts of the vitamin.
I haven't seen any studies describing what happens inside the cell to cobalamin which enters by this mechanism.
This might be particularly relevant when supplementing relatively large amounts of the vitamin.
I haven't seen any studies describing what happens inside the cell to cobalamin which enters by this mechanism.
Last edited:
Creachur
Guest
- Messages
- 51
The book Comprehensive B12: Chemistry, Biochemistry, Nutrition, Ecology, Medicine has graphs sourced from the Quadros et al, 1979 paper showing the formation of AdoCbl from MeCbl and vice versa by human lymphocytes in vitro.
That sounds like an interesting book. Is it open-access or free-access, so I can browse through a copy?
Last edited: