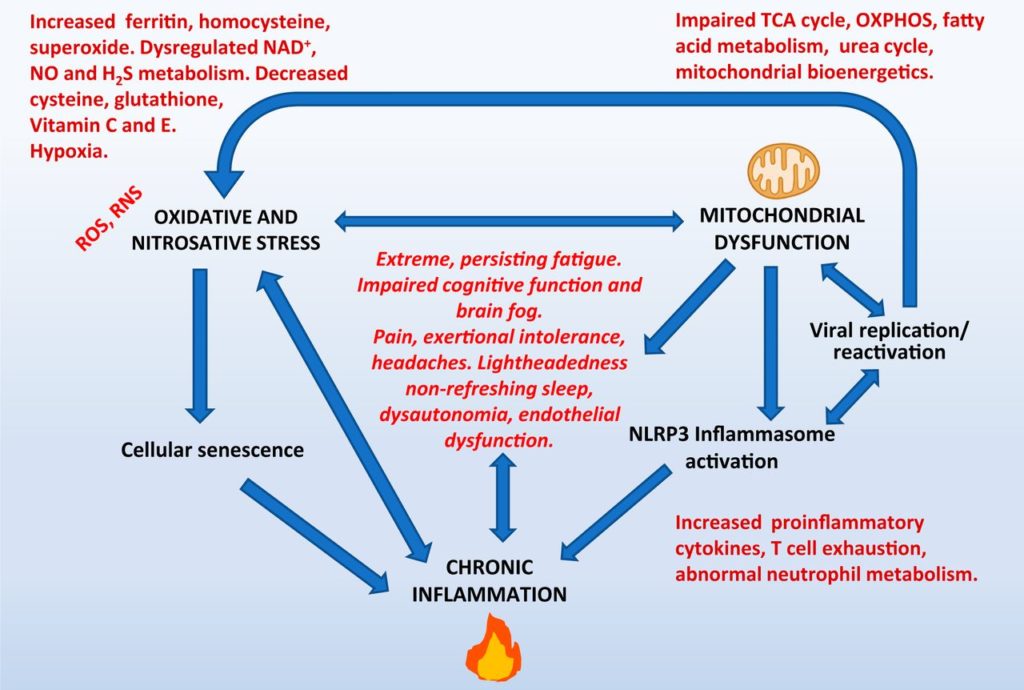
This is an interesting hypothesis. I don't know of much evidence supporting it, but it's interesting.inflammation + gut dysbiosis => (auto)immune response + vascular damage/dysfunction => poor oxygenation, and so on?
This was my (admittedly poor) attempt to capture at least one way in which oxidative stress might lead to vascular damage/dysfunction (in addition to causing direct endothelial damage; Incalza et al., 2018, Vasc. Pharm.). It's based on the ideas of VanElzakker and Proal: peripheral pro-inflammatory cytokines cause an exaggerated "mirror" production of brainstem cytokines (neuroinflammation) which produce chronic low-level sympathetic activation (promoting vasoconstriction) and activate the immune system (which, in some people, exaggerates vasoconstriction due to anti- adronergic/muscarinic receptor antibodies, according to Scheibenbogen's group). (Regarding neuroinflammation, VanElzakker et al., 2018, Fr. Neurol., explains how difficult it is to measure brainstem neuroinflammation, which might explain the Dutch group's recent failure to replicate Nakatomi's early results.)
Of course, oxygen delivery/extraction might be compromised by many factors beyond exaggerated vasoconstriction, including increased vascular stiffness (Bond et al., 2021, JERPH), reduced RBC deformability (Saha et al., 2019, Cl. Hem. Micro.) and even hypercoagulation (to bring it back to the thread topic).