Simon submitted a new blog post:
Part 2: Brain Cells Making us Sick? Messed up microglia could be driving symptoms
Simon McGrath looks at theories that microglia, the brain's immune cells, might be overactive and driving the symptoms of ME/CFS and fibromyalgia.
 Could microglia be behind the symptoms of ME/CFS? Artist's image of a microglia. © 2012 Hagop Kaneboughazian
Could microglia be behind the symptoms of ME/CFS? Artist's image of a microglia. © 2012 Hagop Kaneboughazian
Sickness response is a good thing, helping us survive by resting to fight off infection. But it evolved as a short-term response, and may be harmful if it sets in for the long-term, perhaps playing a role in ME/CFS.
Overactive microglia?
So how could sickness response get stuck in the on position? One possibility is that a chronic infection continues to trigger it, but a chronic infection should exclude an ME/CFS diagnosis.
The last step in the chain, the microglia, are the focus of two other theories which see microglia activated when they should be in their normal, patrolling state.
ME/CFS and fibromyalgia are often referred to as a neuroimmune disease because of the frequent finding of immune dysfunction and widespread symptoms implicating the brain.
Microglia are cells that potentially connect the immune system to the nervous system, responding to immune activation to effectively "turn on" sickness response in the brain. The idea that inappropriate microglial activation could play a major role in ME/CFS is fascinating.
The activation could be due to earlier priming, a severe initial infection, a disturbance in the immune system resulting from problems in the microbiome, autoimmunity or even chronic infection. Microglial activation could be the common endpoint of many different routes to ME/CFS and fibromyalgia.
Microglial research is poised to become a new front in understanding ME/CFS and fibromyalgia, particularly as high-tech tools are developed to probe the state of microglia in the brain.
Dubbo studies: severity of acute illness is key
The Dubbo group in Australia has done research suggesting that in sudden-onset CFS, the severity of the initial infection triggers long-term activation of microglia. They studied patients with infectious mononucleosis or related infections and found that the stronger the initial sickness response, the greater the chances that a patient would go on to develop CFS.
The CFS did not seem to be caused by continuing high cytokine levels. While cytokine levels were initially elevated, they dropped back to normal levels. But, the researchers suggested, the severity of the initial cytokine response might have got the microglia "stuck" in an activated mode, locking the brain into an extended sickness response.
Interferon-gamma, a proinflammatory cytokine that can activate microglia, seemed to play a particularly important role. Patients with two copies of the most active interferon-gamma gene version were likely to be sicker initially and about three times more likely to develop CFS at six months.
This hypothesis got some support from a recent small study which found that microglia in the brains of ME/CFS patients were activated -- exactly what the Dubbo hypothesis predicts.
Microglial 'Priming' theory
 Professor Hugh PerryA second way that microglia could cause a continuing sickness response is by becoming over-reactive. Hugh Perry, Professor of experimental neuropathology from the University of Southampton, and Dr. Jarred Younger of Stanford University have independently proposed that the problem starts with "priming" of microglia by a variety of factors, including previous infections, obesity, chronic stress, and age.
Professor Hugh PerryA second way that microglia could cause a continuing sickness response is by becoming over-reactive. Hugh Perry, Professor of experimental neuropathology from the University of Southampton, and Dr. Jarred Younger of Stanford University have independently proposed that the problem starts with "priming" of microglia by a variety of factors, including previous infections, obesity, chronic stress, and age.
Primed microglia are hypersensitive -- a bit like a primed or armed bomb -- so they react too easily and/or too aggressively, which could lead to long-term sickness response. So an infection or other trigger that would lead to no long-term problems in someone with "normal" microglia could lead to ME/CFS or fibromyalgia in someone with primed microglia.
Primed microglia are measurably different from patrolling ones, with very different receptors on their cell surface. Fewer of the receptors that respond to "keep calm" signals from healthy neurons and more receptors that respond to any "shoot now" signals. The result is an excitable, trigger-happy microglial cell.
Much of the work on priming has been done on animals, since the only sure way to detect priming is to physically examine them. However, studies looking at brains of people who have died of neurodegenerative disease also reveal primed microglia.
Perry believes that priming is an important factor in these illnesses because it could lead to an overly-aggressive immune response, damaging tissue instead of helping it heal.
At the recent International Symposium for CFS/ME in Australia Perry suggested that priming of microglia could be behind this illness as well (though he doesn’t think neurones degenerate in ME/CFS the way they do in the other illnesses he studies). Once primed, Perry argues, an infection could send the microglia into a permanently activated state, triggering a long-term sickness response.
 Dr. Jarred YoungerYounger, a pain researcher at Stanford, independently developed a very similar theory, which he described at the Stanford Symposium and IACFS/ME conference in March. He primarily focused on fibromyalgia in the IACFS/ME talk, but believes the same mechanism could be behind ME/CFS. Priming could lead to the ME/CFS and fibromyalgia in two possible ways, says Younger:
Dr. Jarred YoungerYounger, a pain researcher at Stanford, independently developed a very similar theory, which he described at the Stanford Symposium and IACFS/ME conference in March. He primarily focused on fibromyalgia in the IACFS/ME talk, but believes the same mechanism could be behind ME/CFS. Priming could lead to the ME/CFS and fibromyalgia in two possible ways, says Younger:
However, activation is not the same as priming, and there is currently no way to detect "primed" microglia in living humans. Until there is, there won't really be a way to put this theory to the test. If larger studies show there is consistent activation of microglia, there may well be more attention on the priming hypothesis.
Intriguing treatment possibilities
If microglial activation is a factor in ME/CFS, then it could be treatable -- one reason there is so much interest in this line of research.
Perry and Younger pointed to the potential of microglial inhibitor drugs to calm down the overactive cells or prevent activation in the first place. No microglial inhibitors have been proven to treat ME/CFS or fibromyalgia, but Younger has already run an intriguing small study of low-dose naltrexone (LDN) for fibromyalgia with encouraging results.
The exact mechanism for LDN is uncertain, but Younger has pointed to evidence that at the low doses used naltrexone's main role is probably to inhibit activation of microglia. Other drugs have been shown to inhibit microglial activation in animals, but not yet in humans.
This is definitely an area to watch.
Simon McGrath tweets on ME/CFS research:
Phoenix Rising is a registered 501 c.(3) non profit. We support ME/CFS and NEID patients through rigorous reporting, reliable information, effective advocacy and the provision of online services which empower patients and help them to cope with their isolation.
There are many ways you can help Phoenix Rising to continue its work. If you feel able to offer your time and talent, we could really use some more authors, proof-readers, fundraisers, technicians etc. We’d also love to expand our Board of Directors. So, if you think you can help in any way then please contact Mark through the Forums.
And don’t forget: you can always support our efforts at no cost to yourself as you shop online! To find out more, visit Phoenix Rising’s Donate page by clicking the button below.
 Continue reading the Original Blog Post
Continue reading the Original Blog Post
Part 2: Brain Cells Making us Sick? Messed up microglia could be driving symptoms
Simon McGrath looks at theories that microglia, the brain's immune cells, might be overactive and driving the symptoms of ME/CFS and fibromyalgia.
In Part 1, he described how the body reacts to infection or wounding with a “sickness response” that partly resembles ME/CFS, and how the microglia are the last step in the physiological mechanisms that lead to sickness response.
Could microglia be behind the symptoms of ME/CFS? Artist's image of a microglia. © 2012 Hagop KaneboughazianSickness response is a good thing, helping us survive by resting to fight off infection. But it evolved as a short-term response, and may be harmful if it sets in for the long-term, perhaps playing a role in ME/CFS.
Overactive microglia?
So how could sickness response get stuck in the on position? One possibility is that a chronic infection continues to trigger it, but a chronic infection should exclude an ME/CFS diagnosis.
The last step in the chain, the microglia, are the focus of two other theories which see microglia activated when they should be in their normal, patrolling state.
ME/CFS and fibromyalgia are often referred to as a neuroimmune disease because of the frequent finding of immune dysfunction and widespread symptoms implicating the brain.
Microglia are cells that potentially connect the immune system to the nervous system, responding to immune activation to effectively "turn on" sickness response in the brain. The idea that inappropriate microglial activation could play a major role in ME/CFS is fascinating.
The activation could be due to earlier priming, a severe initial infection, a disturbance in the immune system resulting from problems in the microbiome, autoimmunity or even chronic infection. Microglial activation could be the common endpoint of many different routes to ME/CFS and fibromyalgia.
Microglial research is poised to become a new front in understanding ME/CFS and fibromyalgia, particularly as high-tech tools are developed to probe the state of microglia in the brain.
Dubbo studies: severity of acute illness is key
The Dubbo group in Australia has done research suggesting that in sudden-onset CFS, the severity of the initial infection triggers long-term activation of microglia. They studied patients with infectious mononucleosis or related infections and found that the stronger the initial sickness response, the greater the chances that a patient would go on to develop CFS.
The CFS did not seem to be caused by continuing high cytokine levels. While cytokine levels were initially elevated, they dropped back to normal levels. But, the researchers suggested, the severity of the initial cytokine response might have got the microglia "stuck" in an activated mode, locking the brain into an extended sickness response.
Interferon-gamma, a proinflammatory cytokine that can activate microglia, seemed to play a particularly important role. Patients with two copies of the most active interferon-gamma gene version were likely to be sicker initially and about three times more likely to develop CFS at six months.
This hypothesis got some support from a recent small study which found that microglia in the brains of ME/CFS patients were activated -- exactly what the Dubbo hypothesis predicts.
Microglial 'Priming' theory
Professor Hugh PerryPrimed microglia are hypersensitive -- a bit like a primed or armed bomb -- so they react too easily and/or too aggressively, which could lead to long-term sickness response. So an infection or other trigger that would lead to no long-term problems in someone with "normal" microglia could lead to ME/CFS or fibromyalgia in someone with primed microglia.
Primed microglia are measurably different from patrolling ones, with very different receptors on their cell surface. Fewer of the receptors that respond to "keep calm" signals from healthy neurons and more receptors that respond to any "shoot now" signals. The result is an excitable, trigger-happy microglial cell.
Much of the work on priming has been done on animals, since the only sure way to detect priming is to physically examine them. However, studies looking at brains of people who have died of neurodegenerative disease also reveal primed microglia.
Perry believes that priming is an important factor in these illnesses because it could lead to an overly-aggressive immune response, damaging tissue instead of helping it heal.
At the recent International Symposium for CFS/ME in Australia Perry suggested that priming of microglia could be behind this illness as well (though he doesn’t think neurones degenerate in ME/CFS the way they do in the other illnesses he studies). Once primed, Perry argues, an infection could send the microglia into a permanently activated state, triggering a long-term sickness response.
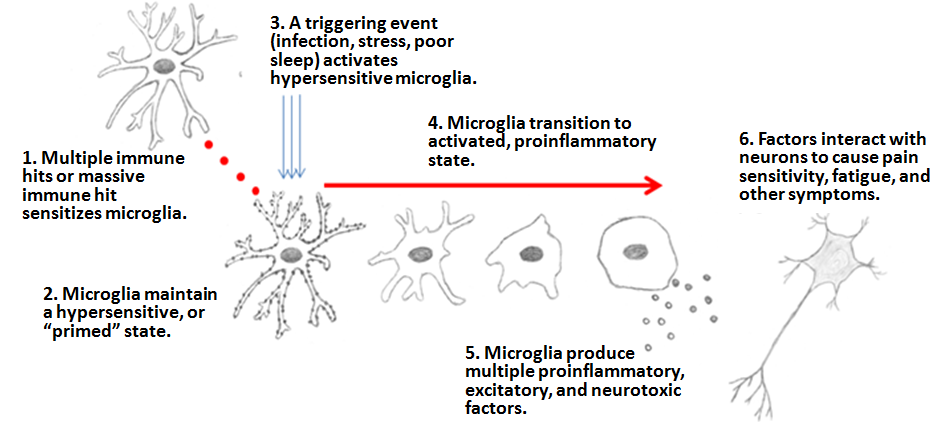
Dr. Jarred Younger- Similar to Perry’s view, microglia might get "stuck" in the activated state: "they fail to revert to their former patrolling state and are therefore constantly producing chemicals that cause ME/CFS symptoms. Microglia are long-lived cells (many, many years), so microglia stuck in [an] state can cause problems for a long period of time."
- Or, primed microglia may react when they shouldn't, producing the flares characteristic of ME/CFS and FM as they become fully activated and pump out the chemicals that cause sickness.
The Microglia Priming hypothesis for fibromyalgia and ME/CFS, © Dr. Jarred Younger. Reproduced with permission.
As yet, there is no hard evidence of microglial activation in fibromyalgia, but that might be about to change: Younger recently reported that a microglial-activation PET study -- similar to the recent one on ME/CFS -- is currently under way at Harvard.
Intriguing treatment possibilities
If microglial activation is a factor in ME/CFS, then it could be treatable -- one reason there is so much interest in this line of research.
Perry and Younger pointed to the potential of microglial inhibitor drugs to calm down the overactive cells or prevent activation in the first place. No microglial inhibitors have been proven to treat ME/CFS or fibromyalgia, but Younger has already run an intriguing small study of low-dose naltrexone (LDN) for fibromyalgia with encouraging results.
The exact mechanism for LDN is uncertain, but Younger has pointed to evidence that at the low doses used naltrexone's main role is probably to inhibit activation of microglia. Other drugs have been shown to inhibit microglial activation in animals, but not yet in humans.
This is definitely an area to watch.
Thanks to @searcher who reported on Dr Younger’s work from Stanford and IACFS/ME, and helped me get the science right in this piece.
Simon McGrath tweets on ME/CFS research:
Phoenix Rising is a registered 501 c.(3) non profit. We support ME/CFS and NEID patients through rigorous reporting, reliable information, effective advocacy and the provision of online services which empower patients and help them to cope with their isolation.
There are many ways you can help Phoenix Rising to continue its work. If you feel able to offer your time and talent, we could really use some more authors, proof-readers, fundraisers, technicians etc. We’d also love to expand our Board of Directors. So, if you think you can help in any way then please contact Mark through the Forums.
And don’t forget: you can always support our efforts at no cost to yourself as you shop online! To find out more, visit Phoenix Rising’s Donate page by clicking the button below.
Last edited: