
I'm taking a Bioinformatics class in Coursera (generally not interesting unless you REALLY want to do mental gymnastics), and one thing we learned about recently is that predictions are made regarding the effects of amino acid substitutions in proteins.
Basically when there is a different variation in a SNP in a gene (such as having an "A" allele instead of "G"), sometimes it results in a missense mutation in the protein created by the gene. That just means that one amino acid, such as cysteine, is substituted with another amino acid, such as glycine.
That can result in the protein behaving much differently, but often has little or no impact. But by looking observing the actual rates of one amino acid being replaced with another (BLOSUM), or by calculating the likelihood of it happening based on the similarities and differences between amino acids (PAM), various matrices (charts) have been created which show the likelihood of specific amino acid mutations occurring.
This also has implications regarding how much effect a mutation is likely to have. If it's a very common one, such as from leucine to isoleucine, it probably won't have much impact due to those two amino acids being extremely similar, and accordingly it'll be fairly common to see such mutations. Whereas glycine behaves very differently than either leucine or isoleucine, and a mutation from glycine to one of those would be much more uncommon.
Here's one such commonly used BLOSUM matrix:
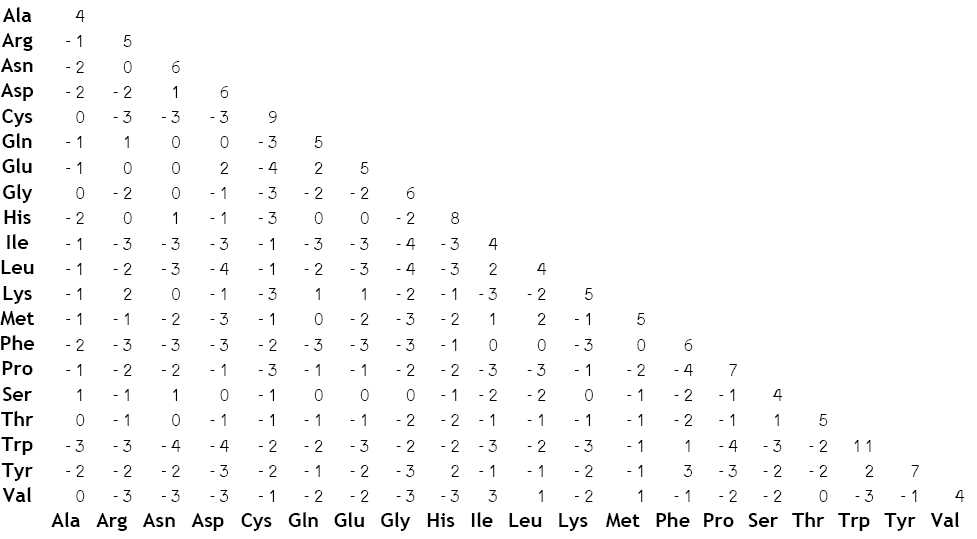
Big numbers mean the mutation is more common, and the most negative numbers mean the mutation is least common.
And here's a chart showing the shared amino acid characteristics used in predicting values for PAM matrices:

Anyhow, it could be relevant in determining how interesting some of our own mutations are
Basically when there is a different variation in a SNP in a gene (such as having an "A" allele instead of "G"), sometimes it results in a missense mutation in the protein created by the gene. That just means that one amino acid, such as cysteine, is substituted with another amino acid, such as glycine.
That can result in the protein behaving much differently, but often has little or no impact. But by looking observing the actual rates of one amino acid being replaced with another (BLOSUM), or by calculating the likelihood of it happening based on the similarities and differences between amino acids (PAM), various matrices (charts) have been created which show the likelihood of specific amino acid mutations occurring.
This also has implications regarding how much effect a mutation is likely to have. If it's a very common one, such as from leucine to isoleucine, it probably won't have much impact due to those two amino acids being extremely similar, and accordingly it'll be fairly common to see such mutations. Whereas glycine behaves very differently than either leucine or isoleucine, and a mutation from glycine to one of those would be much more uncommon.
Here's one such commonly used BLOSUM matrix:
Big numbers mean the mutation is more common, and the most negative numbers mean the mutation is least common.
And here's a chart showing the shared amino acid characteristics used in predicting values for PAM matrices:
Anyhow, it could be relevant in determining how interesting some of our own mutations are